Capítulo 13 - Vasculitis
La vasculitis es un proceso anatomoclínico caracterizado por inflamación y lesión de los vasos sanguíneos. Suele haber afección de la luz vascular asociado con isquemia de los tejidos que reciben flujo del vaso implicado. Tal proceso puede dar lugar a un amplio y heterogéneo grupo de síndromes, porque es capaz de afectar a los vasos de cualquier clase, calibre y localización.
La vasculitis y sus consecuencias pueden ser la principal o la única manifestación de una enfermedad; otras veces, la vasculitis constituye un fenómeno secundario a alguna enfermedad primaria. Además, la vasculitis pueden circunscribirse a un solo órgano, como la piel, o afectar simultáneamente a varios órganos y aparatos.
Una característica importante del conjunto de los síndromes vasculíticos es la gran heterogeneidad y, al mismo tiempo, una considerable superposición entre todos ellos. En general, se supone que la mayor parte de los síndromes de la vasculitis son mediados, al menos en parte, por mecanismos inmunopatogénicos que ocurren por reacción a ciertos estímulos antigénicos, sin embargo, las pruebas que respaldan esta hipótesis son casi en su totalidad indirectas.
13.1 Síndromes de Vasculitis
13.1.1 Síndromes Primarios de Vasculitis
Granulomatosis de Wegener
Síndrome de Churg-Strauss
Poliarteritis nodosa
Poliangitis microscópica
Arteritis de células gigantes
Arteritis deTakayasu
Púrpura de Henoch-Schónlein
Vasculitis cutánea idíopática
Crioglobulinemia mixta esencial
Síndrome de Behçet
Vasculitis aislada del sistema nervioso central
Síndrome de Cogan
Enfermedad de Kawasaki
13.1.2 Síndromes Secundarios de Vasculitis
Vasculitis medicamentosa
Enfermedad del suero
Vasculitis acompañada de otras
enfermedades primarias (Infección,
cáncer, enfermedad reumática).
13.2 Fisiopatologia
Existen tres mecanismos fisiopatológicos de actuación de las vasculitis:
13.2.1 Formación de Depósitos de Complejos Inmunitarios Patógenos
En el mecanismo de la lesión hística en las vasculitis se forman complejos antígeno-anticuerpo con exceso de antígeno que se depositan en las paredes de los vasos, cuya permeabilidad aumenta por la acción de las aminas vasoactivas (histamina, bradicinina y leucotrienos liberados por las plaquetas o los mastocitos) tras mecanismos desencadenados por la IgE. El depósito de los complejos produce activación de los factores del complemento, especialmente de C5a, con un efecto quimiotáctico intenso para los neutrófilos. De inmediato, estas células infiltran las paredes de los vasos, fagocitan a los complejos inmunitarios y liberan enzimas intracitoplásmicas que lesionan la pared vascular. Cuando el proceso se vuelve subagudo o crónico, las células mononucleares infiltran las paredes de los vasos. El denominador común del cuadro resultante es la disminución de la luz de los vasos seguida de lesiones isquémicas en los tejidos cuyo riego sanguíneo depende de los vasos afectados. Diversas variables ayudan a explicar por quéúnicamente algunos tipos de complejos inmunitarios causan vasculitis y sólo algunos vasos se lesionan en cada paciente. Entre ellas estála capacidad del sistema reticuloendotelial para eliminar los complejos circulantes de la sangre, el tamaño y la propiedad fisicoquímica de los complejos inmunitarios, el grado relativo de turbulencia de la sangre circulante, la presión hidrostática intravascular en los distintos vasos y la integridad previa del endotelio vascular.
A este grupo pertenecen:
Púrpura de Henoch-Schónlein.
Vasculitis vinculada a enfermedades vasculares del colágeno.
Enfermedad del suero y síndromes de vasculitis cutánea.
Crioglobulinemia mixta esencial vinculada a hepatitis C.
Poliarteritis nodosa vinculada a hepatitis B.
13.2.2 Anticuerpos Antineutrófilos
Los anticuerpos antineutrófilos citoplásmicos son anticuerpos dirigidos contra ciertas proteínas de los gránulos citoplásmicos de los neutrófilos y monocitos. Estos autoanticuerpos (ANCA) abundan en los pacientes con ciertos síndromes vasculíticos generalizados. La activación de neutrófilos y monocitos a través de ANCA induce la liberación de citocinas pro inflamatorias como IL-1 e IL-8. Pero también se ha demostrado casos en ausencia de ANCA por lo que todavía no se conoce claramente la patogenia de la enfermedad en este subgrupo de vasculitis.
A este grupo pertenecen:
Granulomatosis de Wegener.
Síndrome de Churg-Strauss.
Poliangitis microscópica.
13.2.3 Respuestas Patógenas de los Linfocitos T y Formación de Granulomas
Los complejos inmunitarios pueden inducir respuestas granulomatosas. Las células endoteliales de los vasos pueden expresar las moléculas del antígeno leucocítico humano (human leukocyte antigen, HLA) de la clase II después de ser activadas por citocinas como el interferón (IFN) gamma. Ello permite a estas células participar en las reacciones inmunitarias, como es la interacción con los linfocitos T CD4+, de manera parecida a como actúan los macrófagos que presentan los antígenos. Las células endoteliales pueden secretar IL-1, que es capaz de activar a los linfocitos T e iniciar o propagar los procesos inmunitarios in situ en el interior de la pared vascular. Además, la IL-1 y el TNF-a son potentes inductores de la molécula de adherencia del leucocito al endotelio 1 (endothelial-leukocyte adhesión molecule 1, ELAM-1) y de la molécula de adherencia a las células vasculares 1 (vascular cell adhesión molecule l, VCAM-1), que favorecen la adherencia leucocítica a las células endoteliales de la pared vascular.
A este grupo pertenecen:
Arteritis de células gigantes.
Arteritis de Takayasu.
Granulomatosis de Wegener.
Síndrome de Churg-Strauss.
13.3 Enfermedad de Takayasu
Recibe su nombre por un oftalmólogo japonés (Dr. Mikito Takayasu) que en 1908 publicóun
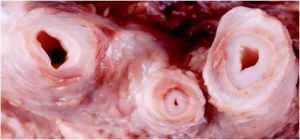
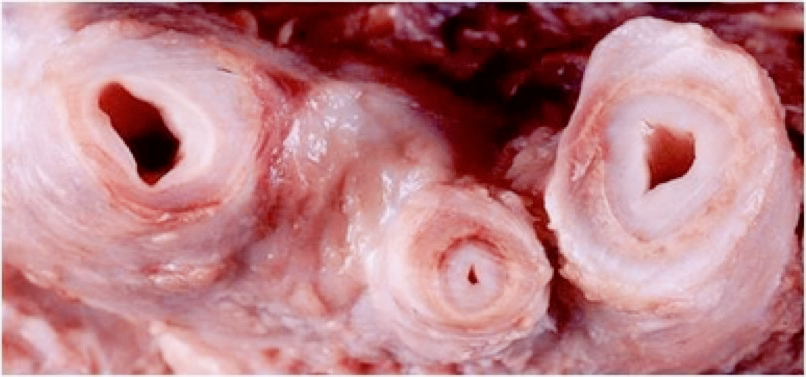
Fig. 1 Aspecto macroscópico de arterias con enfermedad de Takayasu
artículo de una paciente joven donde se describían algunos aspectos que se corresponden a esta enfermedad. La enfermedad de Takayasu es una enfermedad inflamatoria crónica que produce estenosis, obstrucciones y con menos frecuencia aneurismas, preferentemente en la aorta y sus ramas.
13.3.1 Epidemiologia
Afecta entre 1´2 y 2’6 personas por millón de habitantes en la población blanca y 1 de cada 3000 en la asiática. No se tienen datos sobre la incidencia entre población negra.
Es más frecuente en mujeres jóvenes (segunda y tercera décadas de la vida) y niños, especialmente de origen asiático.
13.3.2 Anatomía Patología
La enfermedad de Takayasu es considerada desde el punto de vista anatomopatológico como una vasculitis de células gigantes sin ningún signo histológico patognomónico. Parece comenzar con inflamación de los vasa vasorum, infiltración de la media por linfocitos, granulocitos y demás células que intervienen en el proceso inflamatorio (se han detectado citoquinas como la interleucina 6 y el RANTES) con la destrucción de las fibras de elástica de la capa media y una posterior fibrosis del músculo liso. El proceso acaba afectando a todas las capas de la arteria. La aparición de aneurismas parece relacionarse con procesos evolutivos muy rápidos.
Los pacientes con enfermedad de Takayasu presentan concentraciones elevadas contra células endoteliales lo que parece indicar, junto a su frecuente asociación a ciertas enfermedades reumatológicas, que se trata de una enfermedad autoinmune.
Aunque no existe consenso parece existir cierto componente genético, al menos en pacientes asiáticos, portadores de HLA –B39.2. Tampoco se ha encontrado relación con infecciones, especialmente Mycoplasmas.
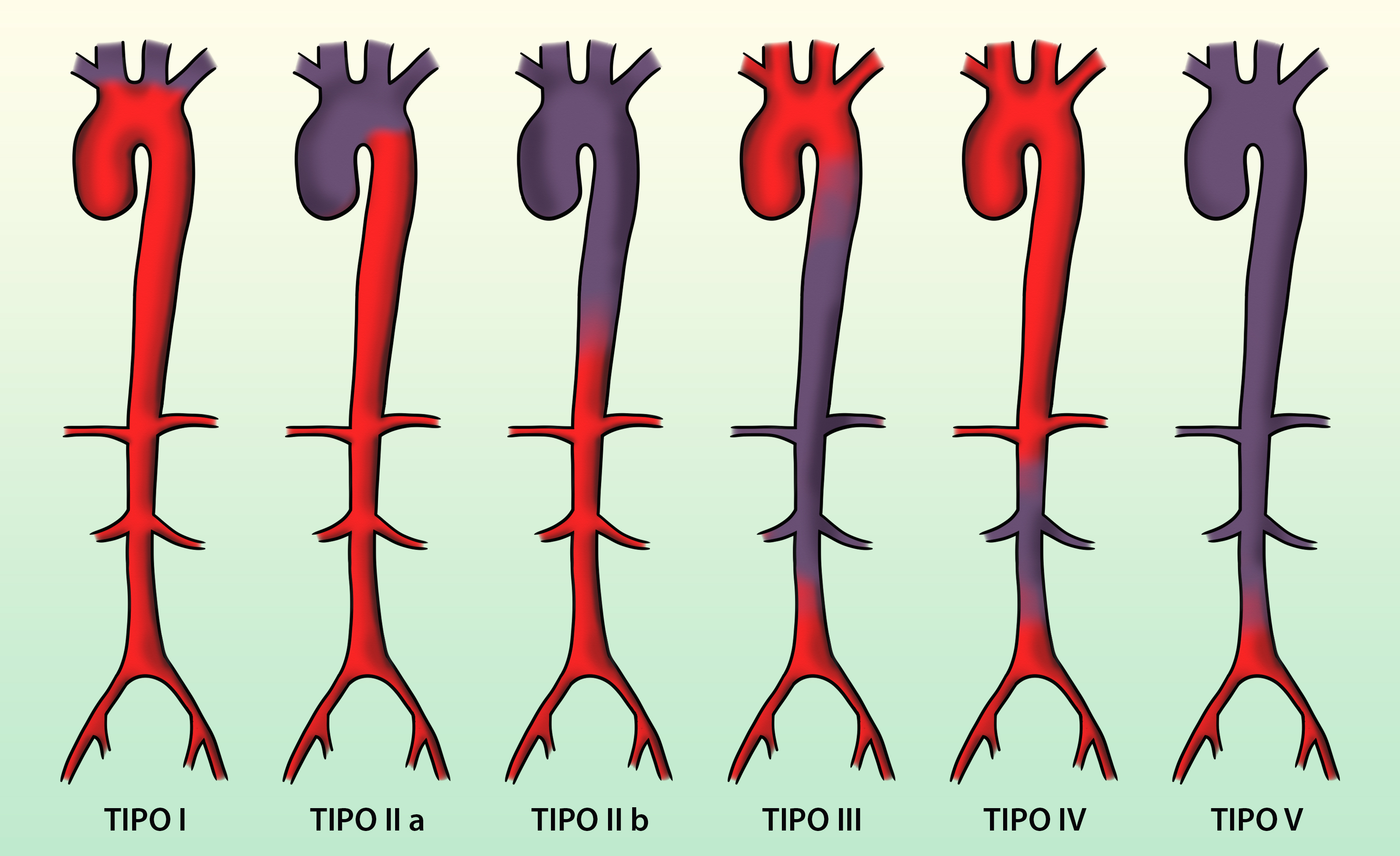
13.3.3 Clasificación
En 1994 la Conferencia Internacional sobre Takayasu establecieron 5 tipos de presentación de la enfermedad:
I: Afectación solo de arterias innominadas, carótidas o subclavias.
IIa: Afectación de aorta ascendente, cayado y grandes vasos.
IIb: Afectación de aorta ascendente, descendente, cayado y grandes vasos.
III: Afectación de aorta descendente, abdominal y sus ramas.
IV: Afectación de aorta abdominal y sus ramas.
V: Afectación de toda la aorta y sus ramas.
Se establece también si en cualquiera de estos grupos existen lesiones coronarias (C+) o de la arteria pulmonar (P+).
Por otra parte, para predecir el pronóstico y gravedad de la enfermedad de Takayasu, se utilizan los criterios pronósticos de Ishikawa, que se basa en 3 factores: 1.- presencia de complicaciones (microaneurismas, HTA grave, insuficiencia aórtica grave y aneurisma aórtico), 2.- la forma de progresión (síntomas crecientes o enfermedad estable) y 3.- la VSG (Velocidad Sedimentación Globular).

Fig. 2 Clasificación de Ishikawa
13.3.4 Formas Clinicas
No se conoce bien la causa de la enfermedad pero el proceso afecta fundamentalmente a la aorta y las grandes arterias. Puede presentarse bajo diferentes formas clínicas. Dado que raramente se puede diagnosticar antes de que se manifiesten las lesiones arteriales, las manifestaciones clínicas dependerán de que vasos estén afectados.
La fase inicial, donde raramente se diagnostica la enfermedad, se caracteriza por malestar general, anorexia, fatiga, adelgazamiento, anemia, mialgias, fiebre y sudores nocturnos en mujeres jóvenes en la segunda y tercera décadas de la vida.
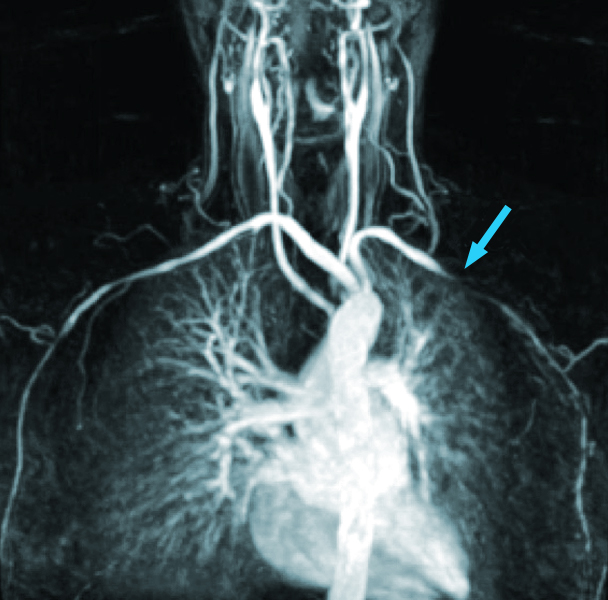
En la segunda fase los síntomas se relacionan con dolor en las zonas donde se produce un fenómeno inflamatorio vascular y, por último, en la tercera fase los síntomas se relacionan con isquemia relacionada con las arterias estenosadas u obstruidas; asíla presentación más frecuente es debida a lesiones arteriales en miembros superiores, especialmente la arteria subclavia izquierda (90%) y puede manifestarse como una simple ausencia de pulsos (73%) o puede presentarse claudicación intermitente en miembros superiores (52%). También es frecuente la afectación de las arterias carótidas y del tronco braquiocefálico. El 20% de los pacientes se manifiestan inicialmente con accidentes cerebrovasculares o cambios visuales, ceguera temporal, mareos o síncopes. Es frecuente la coexistencia de Hipertensión Arterial (HTA) (70%) que en muchos casos estáenmascarada por las estenosis en arterias subclavias. Otra manifestación inicial frecuente es la claudicación intermitente en miembros inferiores, debida a estenosis de la aorta infrarenal. Es frecuente también la aparición de aneurismas ( entre un 30 y 70% de las lesiones aórticas pueden degenerar ) pero no se conoce el riesgo de rotura.
En los pacientes con afectación pulmonar puede aparecer hipertensión pulmonar, disnea de esfuerzo y dolor torácico de tipo pleurítico.
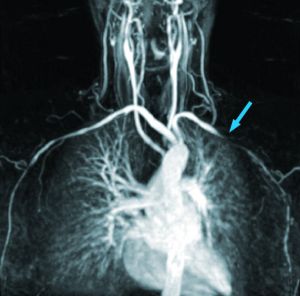
Fig. 3 Afectación de arteria subclavia izda
Se acompaña de lesiones en la piel: entre el 10 y el 20% de los pacientes presenta eritema nodoso (nódulos dolorosos en dermis y tejido subcutáneo), más frecuente en occidentales, o pioderma (dermatitis supurativa caracterizada por la formación de abscesos múltiples) gangrenoso, que es más frecuente en asiáticos. No hay relación entre el sitio de las lesiones de la piel y el vaso afectado, pero las lesiones nodulares suelen aparecer durante la fase aguda de la enfermedad y el pioderma gangrenoso en la fase fibrótica; aparece fenómeno de Raynaud (disminución del flujo normal de la sangre a las puntas de los dedos cuando están expuestos al frío, que se manifiesta con sudoración y frialdad distal en los dedos de manos y pies y coloración azulada o rojiza parcheada de la piel de los dedos) entre el 8 al 14% de los pacientes, generalmente con independencia del compromiso vascular de las extremidades.

13.3.5 Criterios Diagnósticos
Los Criterios de Sharma (1995) son la revisión más reciente para poder diagnosticar esta enfermedad. Para poder decir que un paciente presenta una enfermedad de Takayasu se requiere al menos: dos criterios mayores o uno mayor y dos menores o cuatro criterios menores.
13.3.5.1 Criterios Mayores
1.- Lesiones de la arteria subclavia media izquierda (estenosis severa u oclusión en el segmento medio desde 1 cm proximal al origen de la arteria vertebral hasta un punto localizado a 3 cm del origen de esta arteria).
2.- lesiones de la arteria subclavia media derecha (estenosis severa u oclusión en el segmento medio desde 1 cm proximal al origen de la arteria vertebral hasta 3 cm después).
3.- Signos y síntomas característicos de un mes de duración mínimo (claudicación, disminución /desaparición pulsos, diferencia TA entre brazos > 10 mm Hg, dolor en el cuello, amaurosis fugax, visión borrosa, síncope, disnea o palpitaciones).
13.3.5.2 Criterios Menores
1.- Velocidad de Sedimentación Globular (VSG) elevada (> 20 mm/hora sin otra causa posible).
2.- Sensibilidad carotídea ( a la palpación).
3.- HTA persistente.
4.- Insuficiencia aórtica/enfermedad anular (por auscultación ecocardiografía o angiografía).
5.- Enfermedad arterial pulmonar (oclusión arterial lobular o segmentaria o equivalente).
6.- Lesión en el segmento medio de arteria carótida común izquierda (estenosis grave u oclusión de un segmento medio de 5 cm al menos a 2 cm del origen de la arteria).
7.- Lesión distal en el Tronco Braquiocefálico.
8.- Lesión en la aorta descendente (dilatación estrechamiento, aneurisma o irregularidad luminal).
9.- Lesión en la aorta abdominal (dilatación estrechamiento, aneurisma o irregularidad luminal).
10.- Lesión en las arterias coronarias (documentada mediante angiografía en un paciente < 30 años y sin hiperlipemia ni diabetes).

Fig. 4 Estenosis carotídea
13.3.6 Técnicas Diagnósticas
No existe ninguna prueba de laboratorio específica, la VSG y la Proteína C reactiva, que son marcadores indirectos de actividad inflamatoria, se utilizan como indicadores pronósticos y de respuesta terapéutica.
El diagnóstico debe basarse fundamentalmente en estudios radiológicos.
En las fases iniciales la enfermedad pueden detectarse engrosamientos de la pared de la aorta y la arteria pulmonar y para esto la RNM y el TAC son las herramientas más útiles.
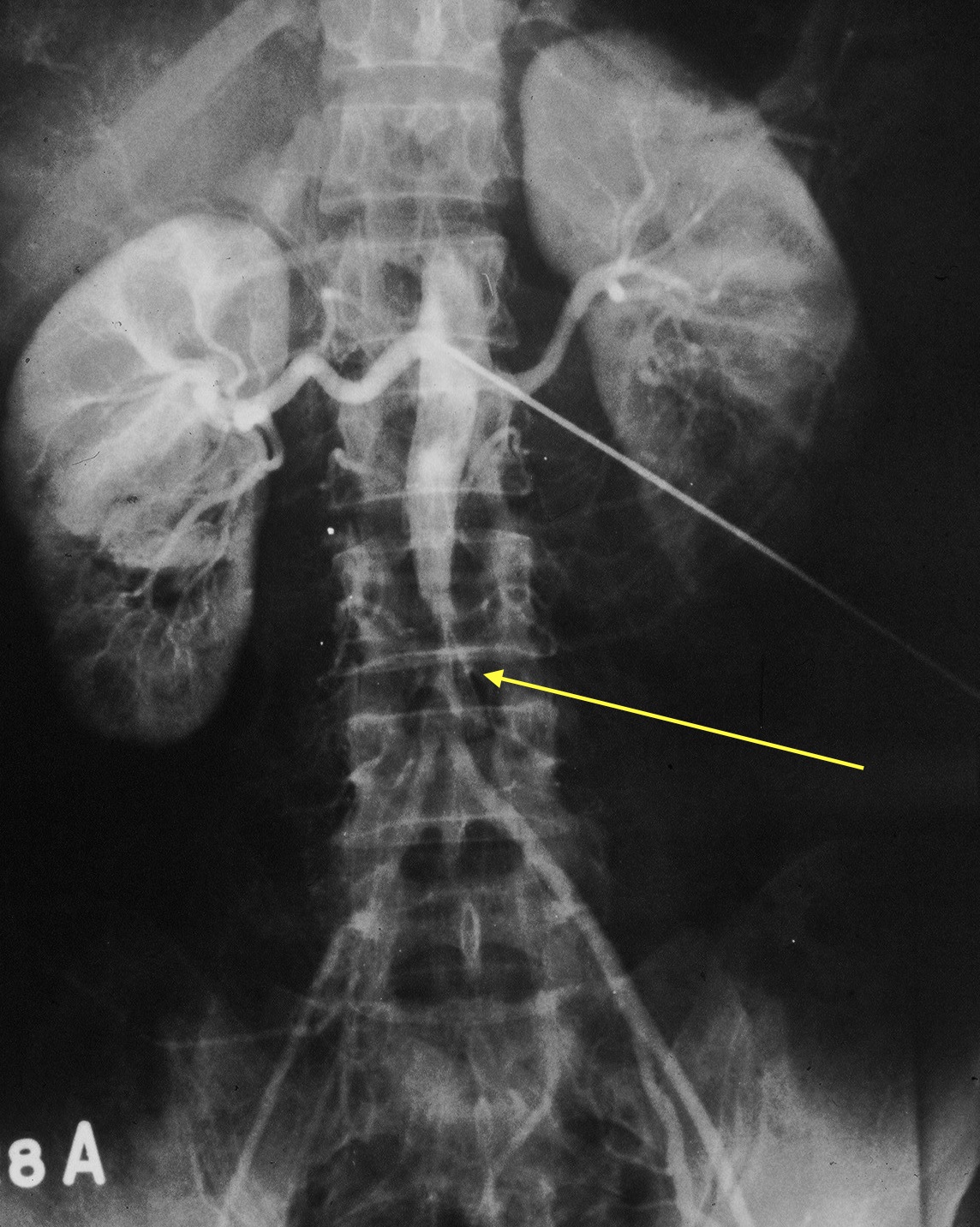
Cuando la enfermedad estádesarrollada y existen manifestaciones clínicas la angiografía es la técnica de elección. Numerosos autores son partidarios de un estudio angiográfico completo cuando se sospecha la enfermedad.

Fig. 5 Oclusión aorta abdominal terminal en mujer joven con Takayasu
La ecografía es una técnica escasamente útil para el diagnóstico y seguimiento de la enfermedad.

13.3.7 Tratamiento
En la fase activa el tratamiento debe ir dirigido contra la respuesta inflamatoria sistémica. En estos casos los esteroides son eficaces aunque en muchos casos la enfermedad se reactiva al suspenderlos.
Existen dos pautas de tratamiento que han mostrado eficacia:
1.- Pauta Ishikawa: Prednisolona (30-50 mg/día) y disminuir la dosis según la VSG. La mayoría de estos pacientes debe continuar durante años con dosis de 15 mg/día.
2.- Protocolo NIH: Prednisolona 1 mg/kg/día. Si no responde a la dosis o no se puede reducir la dosis e interrumpir al cabo de 90 días se añade un citotóxico (ciclofosfamida -2 mg/kg/día-, metrotrexato - 7,5-25 mg/semana- o azatioprina 1-2 mg/kg/día).
La combinación más eficaz y con menores efectos secundarios era con metrotrexato. Si se conseguía remisión de la enfermedad durante 5 años no volvían a recidivar.
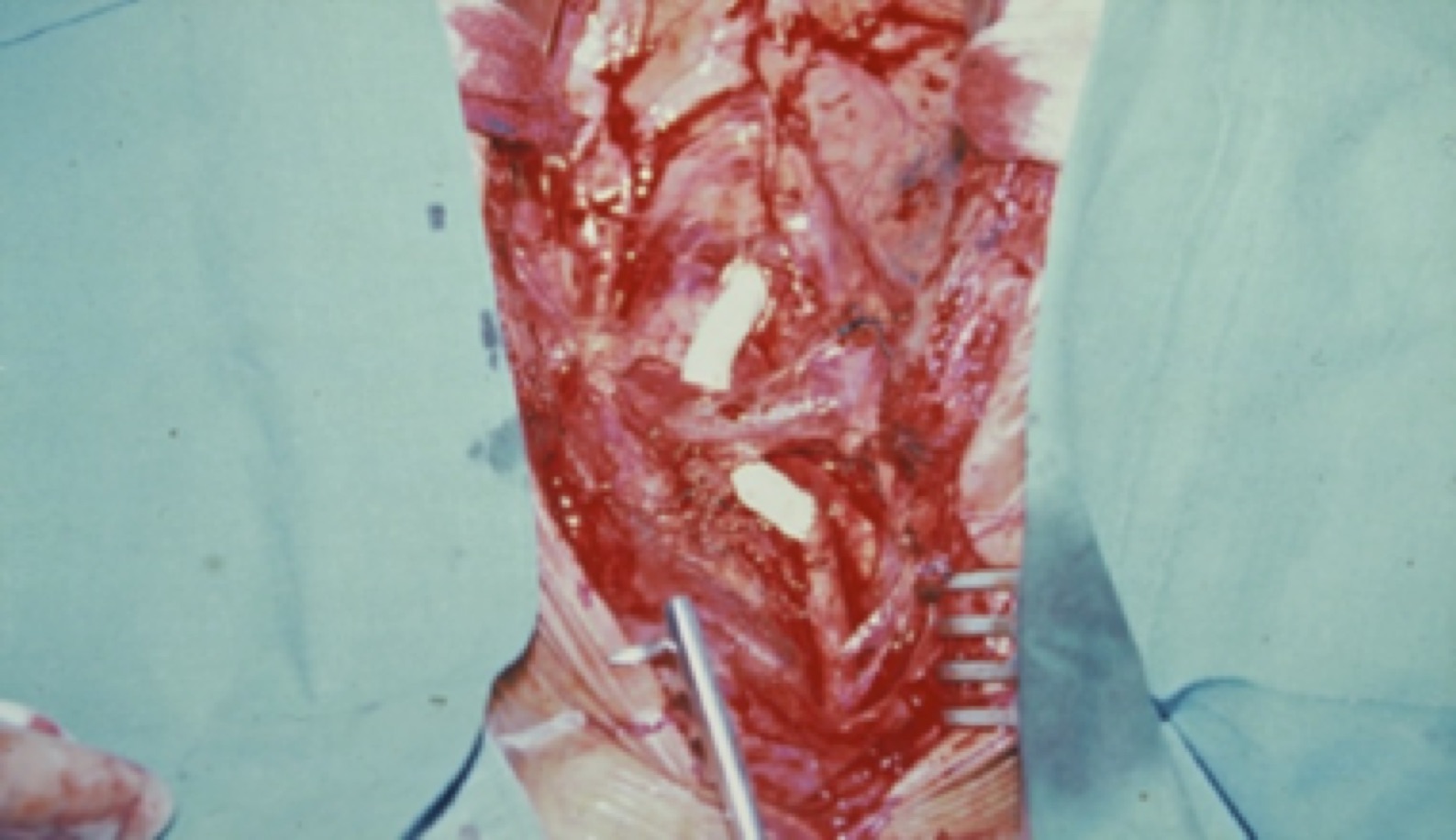
En la fase crónica la cirugía y/o tratamientos endovasculares son las técnicas de elección. Los criterios de indicación son: HTA vasculorrenal, isquemia miocárdica por estenosis coronaria, claudicación invalidante, isquemia cerebral o estenosis de 3 o más vasos cerebrales, insufiencia valvular aórtica, aneurismas torácicos o abdominales > 5cm y severa coartación aórtica. Casi un 30% de los pacientes puede llegar a necesitarla, los resultados son peores cuando se realizan en la fase activa. A pesar de ser pacientes jóvenes los riesgos quirúrgicos son mayores que en la población general ya que el 25% tienen lesiones coronarias, el 60% HTA y casi el 80% tienen fracción de eyección ventricular menor del 45%. Las endarterectomías, angioplastias y stents tienen un alto porcentaje de recidivas y reestenosis sintomáticas. La técnica de elección es el by-pass.
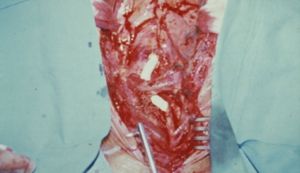
Fig. 6 By-pass aorto carotideo derecho con prótesis PTFE
13.3.8 Pronóstico
La evolución es muy variable y la mortalidad a 5 años, según diversos estudios, varia entre 0 y 35% dependiendo de la existencia o no de lesiones mayores. La muerte se produce fundamentalmente por insuficiencia cardiaca congestiva, rotura de aneurisma, enfermedad cerebrovascular, insuficiencia renal e IAM.
13.4 Arteritis de Células Gigantes o Enfermedad de Horton
Es una vasculitis inflamatoria, crónica y sistémica que afecta sobre todo a arterias de mediano y gran calibre, preferentemente en mujeres mayores de 50 años. También se conoce como arteritis granulomatosa y arteritis de células gigantes. Estáíntimamente relacionada con la polimialgia reumática que aunque puede presentarse de forma aislada, sin arteritis, es muy común que pueda presentarse en el 40 a 50% de los pacientes con arteritis de células gigantes. Además, alrededor de 10 a 20% de los enfermos que en principio presentan manifestaciones de polimialgia reumática aislada más tarde experimentan arteritis de células gigantes. Esta importante relación clínica junto con datos derivados de estudios fisiopatológicos ha apoyado cada vez más que la arteritis de células gigantes y la polimialgia reumática representan cuadros clínicos diversos de un mismo proceso patológico.
13.4.1 Epidemiología
Afecta casi exclusivamente a la raza caucásica y la proporción hombre:mujer es de 1:3. con prevalencia de 18 casos/100.000 habitantes/año, que sube hasta 32/100.000 entre mayores de 50 y a 49/100.00 entre los mayores de 80 años. Es más frecuente en países nórdicos y menos frecuente en los países mediterráneos. Parece existir vinculación con los alelos del locus HLA-DRB1, especialmente la variedad HLA-BDR1*04. Se piensa que existe un factor ambiental desencadenante no identificado. La forma de polimialgia reumática es una variante muy generalizada y cada vez más diagnosticada.
13.4.2 Anatomía Patológica
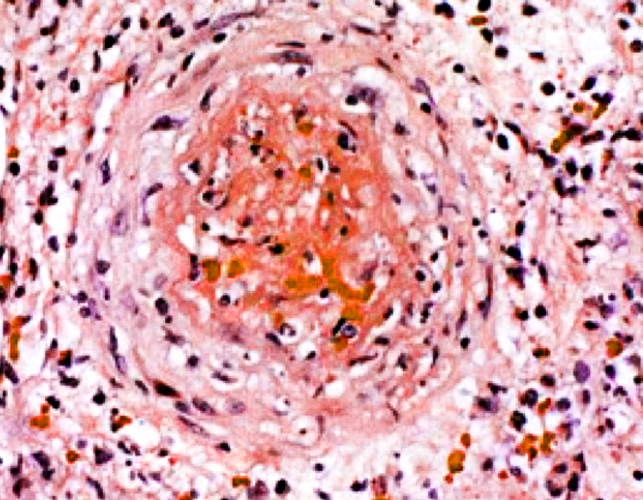
Generalmente se afectan el cayado aórtico y los troncos supraaórticos extracraneales especialmente en las ramas de la carótida externa y la arteria temporal, estando indemnes los vasos intracraneales. Histológicamente se caracteriza por un proceso inflamatorio con invasión de la pared arterial por linfocitos T y macrófagos. La lámina elástica interna estáedematizada y fragmentada y se encuentran depósitos de células gigantes multinucleadas en las fases del proceso. La túnica media es progresivamente sustituida por tejido conjuntivo. Las lesiones son cortas con segmentos arteriales sanos entre las lesiones. Signos histológicos patognomónicos son la fragmentación de la lámina elástica interna y media y las reacciones granulomatosas a de cuerpo extraño.
13.4.3 Clínica
La arteritis de células gigantes suele iniciarse como un síndrome seudogripal: febrícula, cansancio y cefalea (60-98%), posteriormente aparece la claudicación maxilar (40%), las artralgias y dolor en el trayecto de la arteria temporal. Entre el 20 y 60% de los pacientes refieren alteraciones visuales: escotomas, amaurosis fugax, diplopía o pérdida permanente de la visión.
En la variante de polimialgia reumática la clínica se caracteriza por rigidez, dolor sordo en los músculos de cuello, hombros, parte baja del dorso, caderas y muslos en un 60% de los casos.
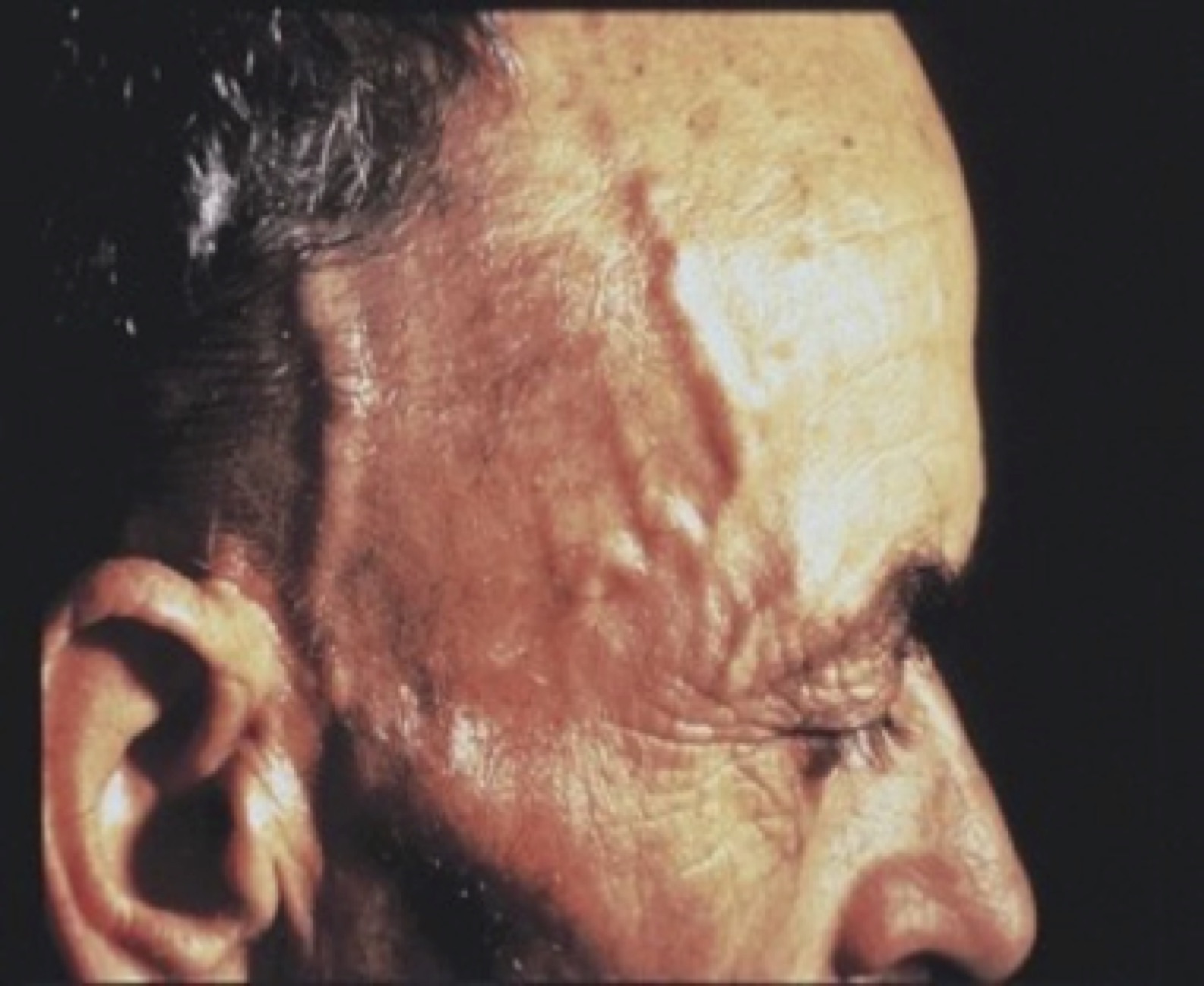
A la exploración puede existir dolor selectivo en el trayecto de las ramas de la carótida externa afectada (temporal, maxilar, facial…) con pulsos palpables en la etapa inicial y pueden apreciarse signos inflamatorios a lo largo del trayecto afectado del vaso (cordón duro, doloroso y eritematoso). Si aparece ceguera suele deberse a la afectación de la arteria oftálmica. Hay que buscar posibles soplos carotídeos.

Fig. 7 Inflamación de arteria temporal
13.4.4 Técnicas Diagnósticas
Los datos analíticos son irrelevantes aunque puede existir VSG (100 mm/h), fibrinógeno y proteína C reactiva elevadas, leucocitosis, anemia normocítica, prolongación del tiempo de protrombina y elevación de transaminasas y fosfatasa alcalina.
RNM con estenosis en ambas arterias subclavias. La exploración con Eco-Doppler permite localizar el segmento afectado del vaso y muestra velocidades de flujo alteradas.
La biopsia de la arteria temporal es el método diagnóstico por excelencia y se debe tomar una muestra de entre 4 y 7 cm.
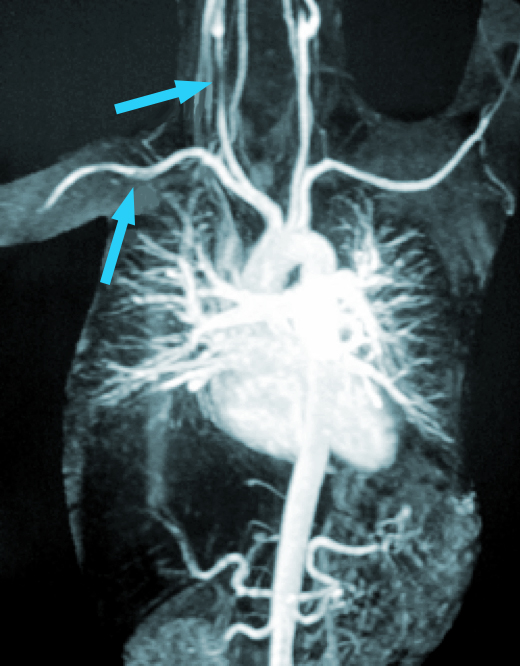
La RNM de alta resolución puede ser de gran ayuda en el diagnóstico al mostrar indicios de engrosamiento y captación de contraste en la pared de los vasos, al igual que las estenosis de los grandes vasos.
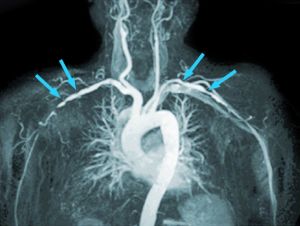
Fig. 8 RNM que muestra lesiones en a. subclavia izquierda
La arteriografía puede pasar por alto muchas lesiones pero cuando se detectan suelen ser estenosis afiladas con dilataciones post-estenóticas, algunos segmentos ocluidos y circulación colateral en aorta, carótidas y vertebrales.
13.4.5 Tratamiento
Dado el riesgo de ceguera aguda, ante la sospecha de arteritis de la temporal, debe iniciarse su tratamiento de forma inmediata con corticoides y posteriormente confirmar el diagnóstico (debe realizarse la biopsia en un plazo no superior a 2-3 días tras el inicio del tratamiento para no tener falsos negativos). La pauta a realizar sería prednisona a dosis de 40-60 mg/día durante un mes, seguida de reducción gradual, pero si no desaparecen los síntomas se debe prolongar el tratamiento generalmente una media de dos años. La recidiva de los síntomas al intentar reducir la dosis es del 60-85% de los casos. La VSG se utiliza como indicador de la actividad inflamatoria y marcador para decidir si se aumentan o disminuyen las dosis de corticoides. Para prevenir complicaciones isquémicas cefálicas se debe asociar ácido acetilsalicílico.
Cuando se presenta polimialgia reumática aislada se utilizan dosis de prednisona menores (10-20 mg /día).
En caso de ceguera se debe administrar metilprednisolona 1 gr/día durante 3 días, aunque solo se consigue la recuperación en un 4-12% de los casos.
Casi nunca se necesita reconstrucción arterial de las lesiones salvo aneurismas, disección aórtica, isquemia persistente de un territorio a pesar del tratamiento e insuficiencia valvular aórtica. Nunca debe realizarse en la fase aguda de la enfermedad.
El pronóstico de la enfermedad es favorable si se instaura tratamiento precoz pero el seguimiento debe ser para toda la vida, especialmente en los casos en que coexiste la arteritis de células gigantes con polimialgia reumática.
13.5 Enfermedad de Buerger
También llamada Tromboangeitis Obliterante es un trastorno inflamatorio, segmentario, no aterosclerótico y que afecta habitualmente a las arterias y venas de pequeño y mediano calibre de las extremidades superiores e inferiores. Afecta a todas las razas aunque es más frecuente en el Medio y Lejano Oriente. En estos países afecta a 5 de cada 100. 000 habitantes, siendo menos frecuente en Europa y EE.UU. Su prevalencia ha disminuido al caer el consumo de tabaco.
13.5.1 Etiologia
Se considera una vasculitis pero con características histológicas e inmunológicas diferenciadas. Su causa es desconocida pero se ha demostrado una estrecha relación entre remisiones, agudizaciones y hábito de fumar. De forma que en los pacientes que abandonan radicalmente el tabaquismo la evolución de la enfermedad suele ser benigna, mientras que en los fumadores se detecta un empeoramiento progresivo. Para evaluar el efecto real del tabaco en las recurrencias de la enfermedad se estárealizando en Japón un estudio epidemiológico a largo plazo evaluando los niveles de la COTININA (el principal metabolito de la nicotina) en orina: se considera fumadores activos a aquellos que presentan niveles superiores a 50 ng/mg de cotinina, fumadores pasivos a aquellos con niveles entre 10-50 mg/mg y no fumadores a los que tienen niveles inferiores a 10 ng/mg. Este estudio debería establecer la relación estadística entre desarrollo y recurrencia de la enfermedad con el grado de exposición al tabaco.
También se ha barajado una posible inmunopatogénesis ya que en estos pacientes se ha detectado en las paredes de los vasos enfermos niveles elevados del factor C4 del complemento, anticuerpos antielastina, anticolágeno, sensibilidad celular al colágeno humano tipos I y III y anticuerpos Ig M, Ig G, Ig A y factor C3. En general se considera que en algunos pacientes existe una disfunción endotelial que pudiera actuar en la formación de trombos asociado a estados de hipercoagulabilidad.
El análisis HLA muestra una frecuencia significativamente alta en los grupos antigénicos Aw24, Bw4O, Bw54, Cwl y DP,2. De todas formas el significado etiológico de estos datos es bastante incierto y de momento generan mas dudas que certezas.
En resumen, el único factor etiológico o estrechamente relacionado como desencadenante de la enfermedad y de su progresión es el tabaco.
13.5.2 Anatomía Patológica
La enfermedad de Buerger es un proceso obstructivo inflamatorio que primariamente afecta tanto a las arterias como a las venas de pequeño y mediano calibre; podemos distinguir dos fases cronológicas:
Fase inicial. Se caracteriza por una inflamación aguda de todas las capas de la pared, especialmente en las venas, y se asocia a trombosis oclusiva. Macroscópicamente la arteria esta tensa y edematizada, al igual que el tejido periarterial. La luz del vaso esta obstruida y la pared muestra signos inflamatorios focales, células gigantes, células epiteloides y leucocitos formando microabscesos y macrófagos gigantes se encuentran en el trombo; en la pared se aprecia proliferación ligera de células
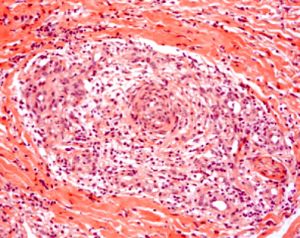
Fig. 9 Fase inicial
intimales con integridad de la lámina elástica interna, linfocitos y fibroblastos infiltran la media y adventicia sin lesiones necróticas. La reacción granulomatosa con células gigantes en el trombo es típica de la fase inicial de la enfermedad de Buerger y nunca se ve en el trombo asociado a la arteriosclerosis. Dos condiciones son necesarias para poder observar células gigantes fagocíticas. 1. poco material soluble (fibrina) y 2. activación local de células mesenquimales.
Fase intermedia.- Se observa una organización progresiva del trombo, a menudo con un infiltrado importante de células inflamatorias en el interior del trombo y en grado muy inferior en la pared vascular.
Fase tardía. Macroscópicamente la arteria estáretraída e indurada. Las arterias y venas adyacentes forman cordones duros difícilmente separables. Microscópicamente hay un engrosamiento fibroso de toda la pared con marcada vascularización de la media. Puede existir en fases avanzadas recanalizaciones parciales del trombo.
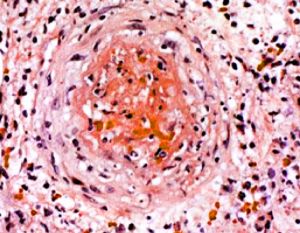
Fig. 10 Fase tardía
Las fases intermedia y tardía expresan poca información diagnóstica pero es casi característico del la enfermedad de Buerger en la fase aguda la marcada proliferación celular y el infiltrado inflamatorio del trombo con integridad de la capa elástica interna, algo que la diferencia de las vasculitis.
13.5.3 Fisiopatología
Lo mas característico es la destrucción de los sistemas de regulación de la microcirculación a causa de las múltiples obstrucciones arteriales desde el mismo inicio de la enfermedad. Esto llevaráa casi todos los pacientes a desarrollar isquemia crítica.
En condiciones normales la velocidad media de la sangre es de 6.8 +/ 1.9 cm/ seg, mientras que en estos enfermos la media es 12 cm/seg. Cuando se asocia una velocidad tan disminuida con índices tobillo/brazo de 0,3, medidos a nivel de los dedos, la aparición de necrosis es casi segura. Si la presión sanguínea en el dedo es menor de 30 mm Hg la curación espontánea de las lesiones es escasa.
Estas características hemodinámicas implicarán la aparición de claudicación plantar a la deambulación. Cuando esta claudicación es gemelar suele estar asociada a obstrucción de las arterias musculares.
13.5.4 Clínica
La forma de presentación clásica de la enfermedad de Buerger es varón joven, fumador y en el que los síntomas se inician antes de los 40-45 años.
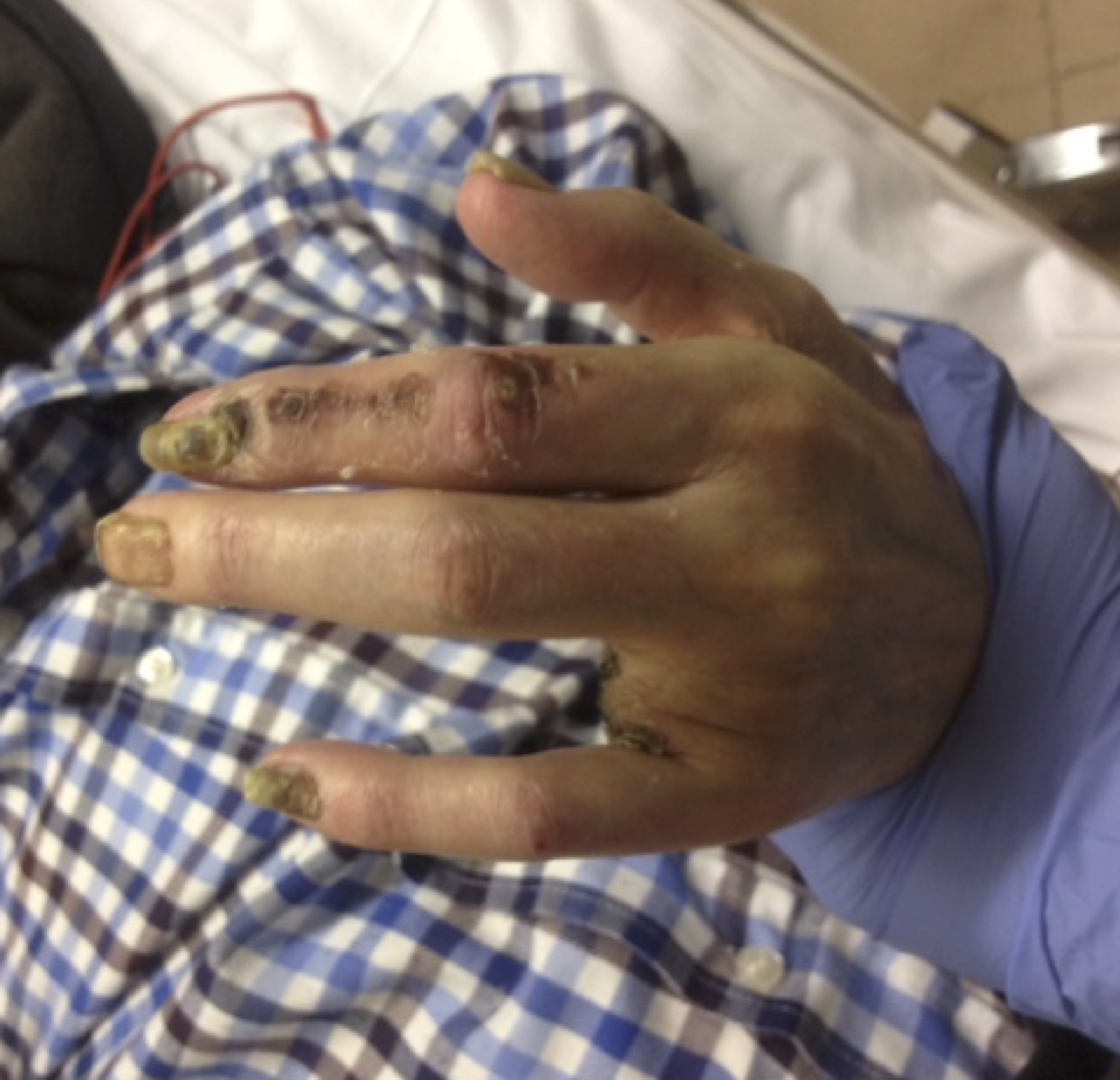
Al desarrollarse la enfermedad en las partes más distales del organismo y con tendencia a la progresión proximal no es extraño que los síntomas iniciales sean progresivos: frialdad, parestesias, cambios cutáneos de coloración, lesiones cutáneas y claudicación. En muchos casos la necrosis y gangrenas anteceden a la claudicación. El dolor de reposo suele localizarse en los dedos.
La clínica sigue dos tipos de evolución: a) progresiva y b) en brotes. Frecuentemente pasan uno o dos años entre el inicio de los síntomas y el inicio de la claudicación gemelar por afectación suprapoplítea, incluso afectando al sector iliaco (afectación infrapoplítea 60%, femoropoplítea 32%, aortoilíaca 8%). Finalmente el curso clínico estámuy influenciado por el tabaquismo, acelerándose el proceso cuando no se cesa en el consumo del tabaco y estabilizándose cuando se abandona.
A la exploración los dedos estarán fríos y húmedos, presentan un aspecto purpúreo y el llenado venoso estáretrasado. Las lesiones necróticas se desarrollan en la punta de los dedos, especialmente el primer dedo, y pueden sufrir infecciones secundarias con dolor intensísimo. No suelen palparse los pulsos tibiales, pero si el poplíteo, aunque éste puede desaparecer en las fases tardías de la enfermedad.
La aparición de flebitis migrans es patognomónica de la enfermedad de Buerger pero suele pasar desapercibida para el paciente en muchos casos.
En resumen podemos decir que clínicamente:
- 98% de los pacientes serán varones.
- La edad de inicio oscila entre 19 y 49 años.
• Las manifestaciones clínicas iniciales son:
. Alteración de la sensibilidad, color o temperatura (40%).
. Úlceras (18%).
. Claudicación plantar (15%).
. Claudicación gemelar (17%).
. Dolor de reposo (10%).
. Tromboflebitis (3%).
Del inicio al final del seguimiento clínico presentarán:
- Úlcera (72%).
- Tromboflebitis (43%)
- Afectación MM. SS. (90%)

Fig. 11 Lesiones tróficas digitales en Enf. De Buerger
13.5.5 Diagnóstico
El diagnóstico de la enfermedad de Buerger se basa en cinco criterios clínicos:
1. Historia de tabaquismo
2. Inicio de los síntomas antes de los 50
3. Lesiones arteriales oclusivas
4. Tanto afectación de la extremidad superior como flebitis
5. Ausencia de otros factores de riesgo
13.5.6 Estudios Diagnósticos
El diagnóstico puede establecerse por la historia clínica y la exploración, sin embargo para establecer el grado de isquemia serán precisos otros estudios:
Exploraciones hemodinámicas no invasivas. Son útiles la medición de la presión sanguínea en tobillo y dedos mediante la fotopletismografia. También es útil la medición de la velocidad de flujo arterial digital. La medición de la P02 subcutánea es buen parámetro clínico y evolutivo de la enfermedad.
Arteriografía, AngioTAC y AngioRNM. Solo sirven para corroborar radiológicamente los hallazgos clínicos y topografiar las lesiones. Los criterios radiológicos diagnósticos serían: lesiones en pequeños vasos, segmentarias, más graves mientras más distal es el vaso, colaterales en tirabuzón o sacacorchos, arterias proximales sin lesiones ateroscleróticas y ausencia de fuentes emboligenas. Nunca se encuentran calcificaciones en las paredes arteriales.

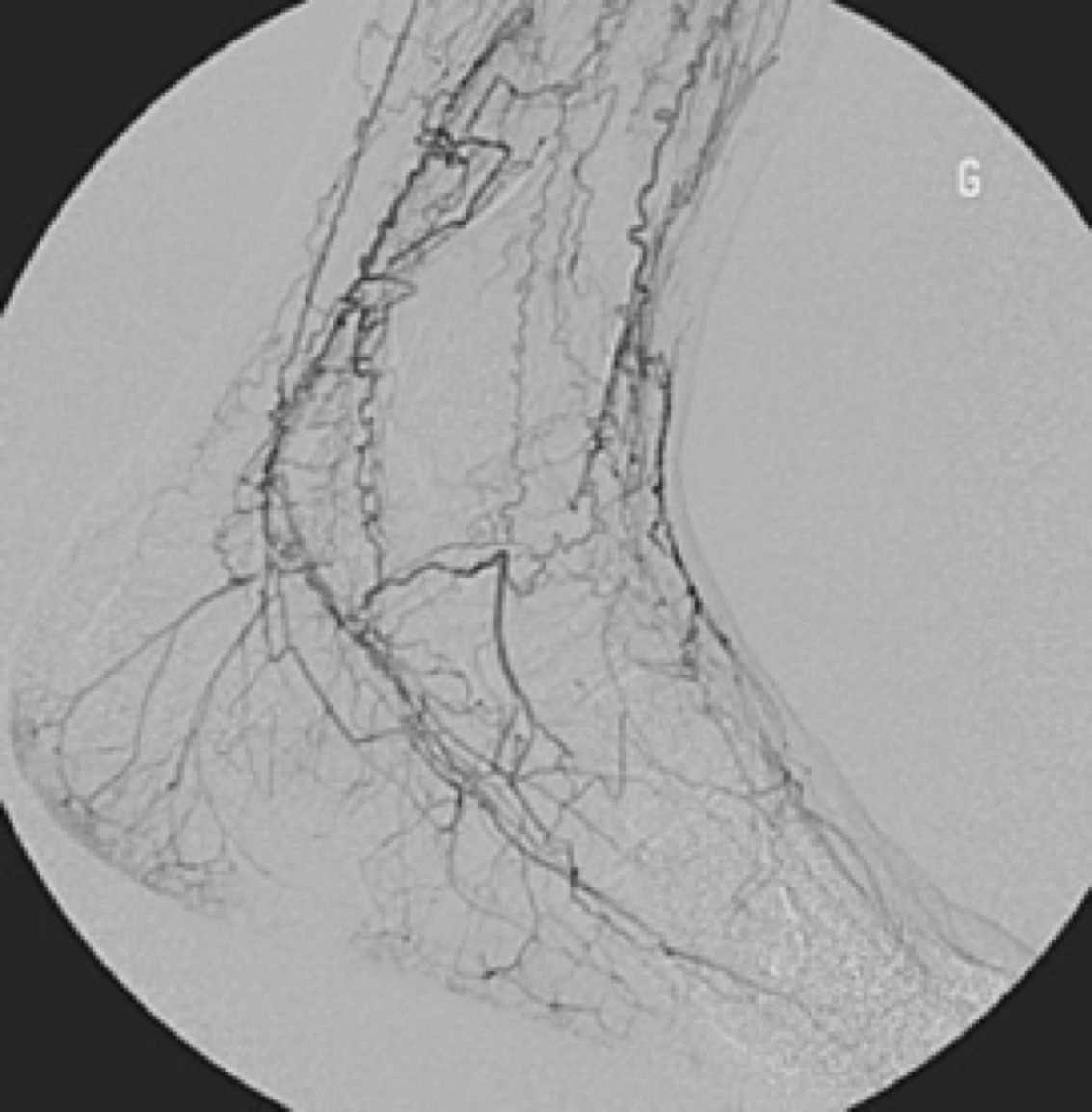
Fig. 12 Típicas arterias en tirabuzón sustituyendo las arterias ocluidas
Analítica. No existen pruebas específicas de laboratorio para diagnosticar la enfermedad de Buerger pero se debe pedir un estudio serológico completo para descartar otro de tipo de patologías: Hemograma, pruebas de función hepática, función renal, glucemia, VSG, proteína C reactiva, anticuerpos antinucleares, factor reumatoide, complemento, determinación de marcadores serológicos del CREST y esclerodermia (ac anticentrómero y SLC 70) y estudio de trombofilia.
13.5.7 Diagnostico Diferencial
En los varones que se sospeche la enfermedad después de los 50 años hay que estudiarlos muy detenidamente para no confundirlos con un proceso aterosclerótico o embolígeno.
Cuando los síntomas se presentan en mujeres hay que realizar el diagnóstico diferencial con enfermedades del colágeno: lupus sistémico, CREST, artritis reumatoide, síndrome mixto del colágeno, síndrome de anticuerpos antifosfolípidos y esclerodermia.
En los pacientes con Takayasu u Horton las lesiones son proximales y no suelen plantear dudas.
Los estudios de imagen también nos permiten diferenciar claramente el síndrome de atrapamiento de la poplítea y la degeneración quística adventicial.
La historia clínica exhaustiva nos permitirádescartar el consumo de ergotamínicos, igualmente el consumo de cannabis, cocaina y anfetaminas pueden provocar lesiones isquémicas similares y es necesario pedir niveles de estos productos para detectar su consumo.
13.5.8 Tratamiento Médico
Se ha demostrado que el abandono del consumo de tabaco es la única forma de detener la progresión de la enfermedad y evitar futuras amputaciones.
Para mejorar el desarrollo de las colaterales no se ha observado que los vasodilatadores produzcan ningún beneficio. La medicación antitrombótica, anticoagulante y fibrinolítica pueden ser útiles para frenar la extensión trombótica en las fases agudas pero su eficacia usadas a largo plazo no está probada.
En los brotes agudos o cuando existen lesiones necróticas es eficaz la utilización de Prostaglandinas (PGE,) por vía IV (510 ng/kg/min) o IA (0.06 ng/ kg/min). Con este fármaco obtenemos una gran vasodilatación y un potente efecto antiagregante plaquetario local y su administración parece ser mas eficaz por la vía intrarterial.
La anestesia epidural ayuda al control del dolor en las fases agudas y a permitir posterior cicatrización de las úlceras.
Un intento reciente de controlar el dolor a largo plazo se basa en la utilización de estimuladores epidurales.
Existen algunas publicaciones que parecen demostrar un efecto beneficioso de la terapia angiogénica con transferencia genética de phVEGF165 para tratamiento de casos críticos de Buerger.
13.5.9 Tratamiento Quirúrgico
1. Reconstrucción arterial directa. Frecuentemente imposible debido a la localización y extensión de las lesiones. En las lesiones infrainguinales es recomendable la utilización de material autólogo, preferentemente la vena safena interna.
2. Simpatectomía. Produce una liberación de los factores vasocronstrictores nerviosos en la parte más distal de la extremidad, especialmente a nivel cutáneo. Estáindicada para facilitar la curación de lesiones tróficas y no suele ser eficaz sobre la claudicación.
3. Desbridamiento. Son necesarios para la curación de las lesiones cutáneas y ulceraciones.
4. Amputaciones. Dado que casi todos estos enfermos presentarán gangrenas o ulceraciones digitales es frecuente la necesidad de amputaciones localizadas. En los pacientes que realizan un adecuado tratamiento no se necesitan amputaciones mayores pero en casos avanzados es necesario recurrir a amputaciones supra e infracondílea.
5.- Otras opciones como tratamientos angiogénicos o estimuladores epidurales despiertan gran interés pero precisan evaluaciones mas exhaustivas.
13.6 Fibrodisplasia Arterial
Se trata de un grupo heterogéneo de trastornos aneurismáticos y oclusivos de origen no aterosclerótico.
Las principales formas son:
1.- fibrodisplasia de la íntima
2.- hiperplasia de la media
3.- fibrodisplasia de la media
4.- displasia perimedial
Los trastornos displásicos afectan a los siguientes vasos: arterias renales, arterias cerebrales intra y extracraneales, arterias axilares, subclavias y braquiales, tronco celíaco, arteria mesentérica superior , inferior y sus ramas, arterias ilíacas, femorales, poplíteas y tibiales y finalmente la aorta.
También puede afectar a venas superficiales de la extremidad inferior y a la vena renal. En las arterias pulmonares es excepcional.
En la mayoría de los casos la displasia fibromuscular representa una arteriopatía sistémica.
13.6.1 Arteria Renal
Es el vaso que más frecuentemente se afecta por la displasia fibromuscular. Su incidencia en la población general se considera <0’5%.
Fue descrita por primera vez en 1938 y es la segunda causa de HTA corregible quirúrgicamente.
Afecta esencialmente a mujeres y se ha especulado con algún factor genético asociado (no demostrado en la actualidad) o la influencia hormonal sobre las paredes de los vasos. También se especula sobre la influencia mecánica de la ptosis renal en la aparición de estas lesiones. También existe una teoría isquémica de la pared de la arteria ya que los vasos en que asienta principalmente tienen escasos vasa vasorum. Afectan fundamentalmente al riñón derecho.

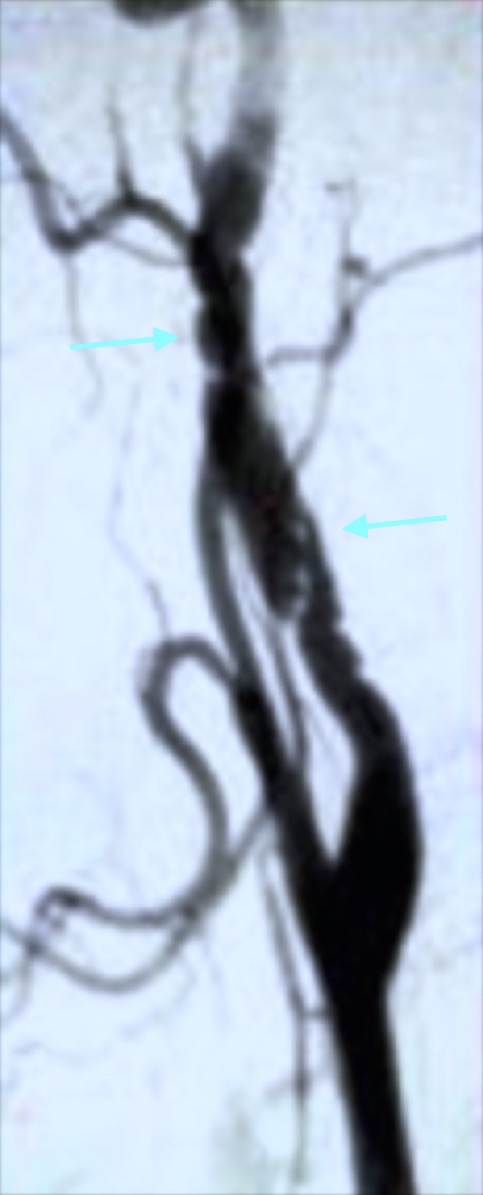
Fig. 13 Arteriografía. Fibrodisplasia renal
13.6.1.1 Fibrodisplasia de la Íntima
Afecta por igual a varones y mujeres. Supone el 5% de todas las estenosis displásicas renales.
Es más frecuente en niños, adolescentes y jóvenes.
Es unilateral y se presenta en el tronco principal de arteria renal como lesión estenótica lisa y focal. Raramente se presenta con lesiones segmentarias.
La fibrodisplasia primaria de la íntima se caracteriza por unas células mesenquimales subendoteliales organizadas irregularmente en el seno de una matriz laxa de tejido conjuntivo fibroso y que protruyen hacia la íntima. La lamina elástica estásiempre presente. La media y adventicia son normales. No se detectan células espumosas o inflamatorias. La lesion es circunferencial.
Se desconoce la etiología aunque algunos hallazgos y teorías apuntan a una forma de arteritis curada secundaria a proceso infeccioso-inmunológico.
La progresión de la lesión parece deberse sobre todo a las alteraciones hemodinámicas producidas por la estenosis.
13.6.1.2 Hiperplasia de la Media
Es poco frecuente en arterias renales (<1%) y se da mas en mujeres en la cuarta y quinta década de la vida.
Se presenta como estenosis focales en el tercio medio de la arteria renal.
Se caracteriza por aumento de células musculares lisas poco desorganizadas y con escasa sustancia fundamental. La íntima y adventicia son normales.
No existe etiología clara.
13.6.1.3 Fibrodisplasia de la Media
Representa el 85% de las lesiones vasculorrenales displásicas. En el 90% son mujeres y es rara en la raza negra.
Este tipo solo se encuentra en arterias renales, carótida interna extracraneal e ilíaca externa.
Su presentación puede variar desde una lesión focal estenótica a su forma más frecuente: un rosario de estenosis y evaginaciones aneurismáticas. Esta forma nunca se presenta antes de la menarquia.
Suelen ser lesiones bilaterales (60-70%) y afectar al tercio distal de la arteria renal y, a veces, a ramas segmentarias de primer nivel (25%). Hasta en dos terceras partes de los pacientes las lesiones progresan con el tiempo.
Se conocen dos formas histológicas: la forma periférica afecta a la media externa y la forma difusa afecta a toda la media. Se caracteriza la forma periférica por aparición de tejido conjuntivo fibroso y compacto que va sustituyendo al músculo liso de la capa externa de la media. No afecta a la íntima ni a la lámina elástica interna.
La forma difusa se caracteriza por una marcada desorganización del músculo liso con distintas formas de fibrosis. Existe fragmentación de la lámina elástica interna y fibrosis subendotelial. En formas extremas pueden aparecer disecciones.
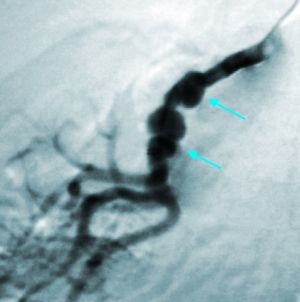
Fig. 14 Fibrodisplasia de la media
13.6.1.4 Displasia Perimedial
Puede coexistir con la fibrodisplasia de la media y muchos consideran que ambas forman parte de una misma enfermedad.
Se presenta como estenosis focales o múltiples sin aneurismas en tercio medio de la artera renal. Su característica específica es la excesiva acumulación de tejido elástico en la union entre la media y la adventicia. Se observa acúmulos de sustancia fundamental y elementos fibrótico.
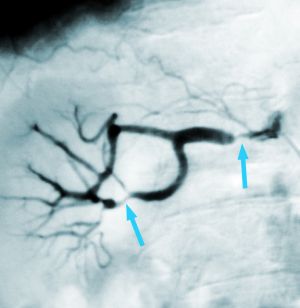
Fig. 15 Displasia perimedial
13.6.2 Arterias Carótidas y Vertebrales
Afectan a <0’4% de la población general y su importancia es potencialmente importante pero cuestionable en la práctica clínica.
Se presenta bajo dos formas
1.- Fibrodisplasia de la íntima
2.- Fibrodisplasia de la media
La primera suele producer elongaciones, acodamientos y bucles en las carótidas.
La segunda se presenta en mujeres, de más de 50 años en el momento del diagnostico, afecta a segmentos de 3-6 cm de longitud con múltiples estenosis. Suelen ser bilaterales. Cuando afectan a las arterias vertebrales suele ser en la parte proximal. En el 25% de los pacientes coexisten lesiones en arterias renales. Entre el 12-25% se asocian a aneurismas cerebrales.
La sintomatología se asocia a progresión de las lesiones que se complican con: estenosis que reducen flujo sanguíneo cerebral, acumulación de trombos que pueden embolizar y a la disección y rotura de la arteria con la formación de fístulas A-V. En general aparecen en menos del 10% de los pacientes.
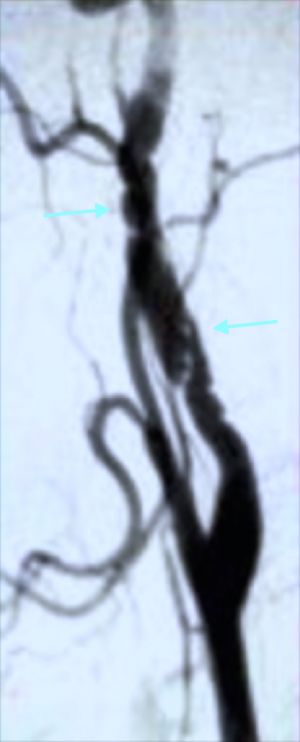
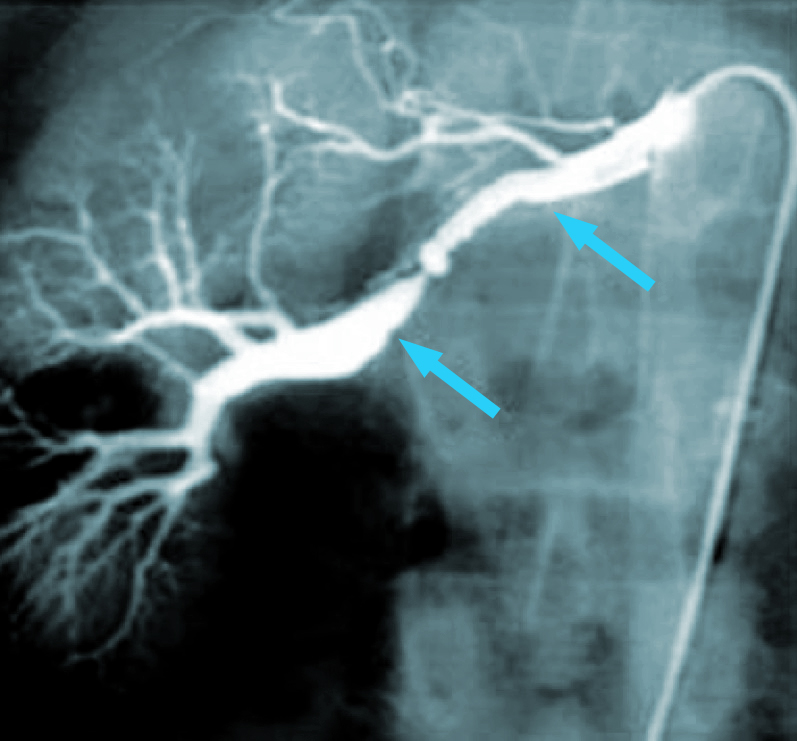
Fig. 16 Arteriografía. Fibrodisplasia carótida interna
13.6.3 Arterias Ilíacas, Femorales , Poplíteas Y Tibiales
Tras arterias renales y carótida interna, la ilíaca externa es el tercer vaso que más se afecta.
Se desconoce su incidencia en la población general pero aparece entre el 1 y el 6% de los pacientes con displasia renal.
Se presenta con estenosis sucesivas intercaladas por aneurismas en el tercio proximal de la arteria. Su forma anatomopatológica es similar a la de arterias renales.
La sintomatología se relaciona con el grado de estenosis o con embolizaciones a miembros inferiores. No es frecuente la disección.
13.6.4 Arterias Axilar, Subclavia Y Braquial
En estos vasos la lesión más frecuente es la fibrodisplasia de la íntima.
Se da algo más en mujeres que en varones. Se asocia a arteritis, traumatismos o alteraciones hemodinámicas.
En general son muy poco frecuentes y no se puede establecer su incidencia ni grupos especiales de presentación.
13.6.5 Arterias Esplácnicas
Puede afectar a tronco celíaco, mesentérica superior e inferior.
No se conoce la etiología y clínicamente puede manifestarse como angor intestinal.
Cuando afecta a mujeres es más frecuente la forma de fibrodisplasia intimal. La fibrodisplasia de la media es poco frecuente en este territorio vascular.
Se presentan como estenosis próximas al origen de estos vasos y es frecuente encontrar dilataciones post-estenóticas.
13.7 Arteritis por Radiación
Se puede desarrollar una arteritis tardía como secuela de la radioterapia en neoplasias.
A menudo se produce un periodo de latencia de al menos diez años antes de las manifestaciones clínicas.
Puede afectarse cualquier vaso que esté situado en el campo de la zona radiada.
Los daños vasculares inducidos por radiación provocan cambios ateromatosos prematuros indistinguibles de otras causas de aterosclerosis.
Histológicamente la arteritis por radiación se caracteriza por una lesion de los vasa vasorum con necrosis isquémica de la pared vascular, fibrosis de la lamina elástica interna, depósitos de fibrina, engrosamiento adventicial y de toda la pared arterial con estenosis de la luz.
Radiológicamente se pueden encontrar lesiones estenóticas focales o estenosis difusas y oclusión y hay que hacer el diagnóstico con otras arteritis que afectan a grandes vasos y con la invasión tumoral de la pared arterial.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
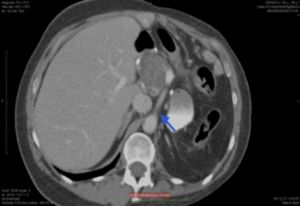
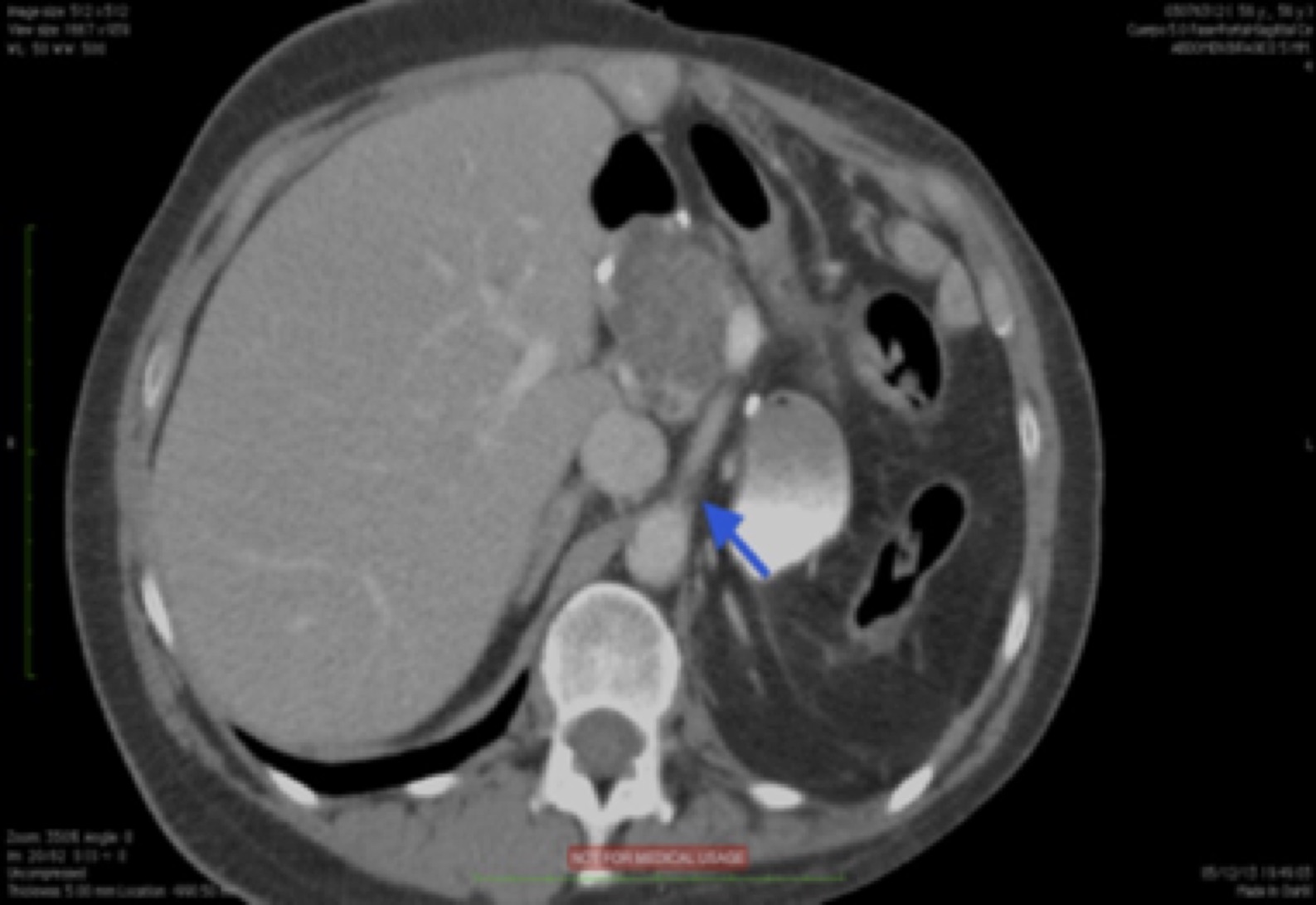
Fig. 17 AngioTAC. Estenosis rádica de origen AMS
Las manifestaciones clínicas son similares a las lesiones ateroscleróticas y dependen de la localización y extension del daño vascular: claudicación, hipertensión, angor intestinal o accidentes cerebrovasculares.
Su tratamiento puede variar desde una reconstrucción vascular abierta hasta un procedimiento endoluminal con endoprótesis. Las complicaciones post-operatorias más frecuentes son infecciones tardías y la difícil cicatrización de las incisiones quirúrgicas.
13.8 Bibliografía
1. Medicina Vascular. Complemento de Braunwald. Tratado de Cardiología. 2ª Edición. Mark A. Greager, Joshua A. Beckman. Edt Elservier Saunders 2013.
2. Rutherford´s Vascular Surgery. 8Th Edition. Rutherford. Jack L. Cronenwett MD, K. Wayne Jhonston. Elservier Saunders 2014.Mujica Pacheco E, Elseid Abukassem K, Alonso Gómez N, Lodeiro Sanz C, Abarrategui C, España Caparrós G. Presentación atípica del fenómeno de Raynaud primario severo. Avances en Cirugía Vascular 2010;1-6. (www.esteve.com)
3. Gallagher K, Clarke M, Scovell S. ascular arteritides in women. J Vasc Surg 57:18S-26S. 2013
4. Maladies Arterielles Non atherosclereuses de l’adulte. E. Kieffer, P. Godeau . Editions AERCV. 1994. ISBN 2-907232-08-8
5. Evans JM, O’Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med 1995;122:502–507.
6. Arend WP, Michel BA, Bloch DA, Hunder GG, Calabrese LH, Edworthy SM, Fauci AS, Leavitt RY, Lie JT, Lightfoot RW Jr., Masi AT, McShane DJ, Mills JA,mStevens MB, Wallace SL, Zvaifler NJ. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum 1990;33: 1129–1134.
7. The International Study Group for Behcet’s disease. Evaluation of diagnostic (’classification’) criteria in Behcet’s disease: towards internationally agreed criteria. Br J Rheumatol 1992;31:299–308.
8. Goie The HS, Steven MM, van der Linden SM, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis: a comparison of the Rome,NewYork and modified New York criteria in patients with a positive clinical history screening test for ankylosing spondylitis. Br J Rheumatol 1985;24:242–249.
9. Gornik HL, Creager MA. Aortitis. Circulation 2008;117:3039–3051.
10. Pipitone N, Versari A, Salvarani C. Role of imaging studies in the diagnosis and follow-up of large-vessel vasculitis: an update. Rheumatology (Oxford) 2008;47:403–408.
11. Restrepo CS, Ocazionez D, Suri R, Vargas D. Aortitis: imaging spectrum of the infectious and inflammatory conditions of the aorta. Radiographics 2011;31:435–451.
12. James OG, Christensen JD,Wong TZ, Borges-Neto S, Koweek LM. Utility of FDG PET/CT in inflammatory cardiovascular disease. Radiographics 2011;31:1271–1286.
13. Gravanis MB. Giant cell arteritis and Takayasu aortitis: morphologic, pathogenetic and etiologic factors. Int J Cardiol 2000;75 Suppl 1:S21–S33; discussion S35–S36.
14. Lane SE,Watts R, Scott DG.Epidemiology of systemic vasculitis. Curr Rheumatol Rep 2005;7:270–275.
15. Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med 2002;347:261–271.
16. Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Limitations of therapy and a guarded prognosis in an American cohort of Takayasu arteritis patients. Arthritis Rheum 2007;56:1000–1009.
17. Hoffman GS, Cid MC, Hellmann DB, Guillevin L, Stone JH, Schousboe J, Cohen P, Calabrese LH, Dickler H, Merkel PA, Fortin P, Flynn JA, Locker GA, Easley KA, Schned E, Hunder GG, Sneller MC, Tuggle C, Swanson H, Hernandez-Rodriguez J, Lopez-Soto A, Bork D, Hoffman DB, Kalunian K, Klashman D, Wilke WS, Scheetz RJ, Mandell BF, Fessler BJ, Kosmorsky G, Prayson R, Luqmani RA, Nuki G, McRorie E, Sherrer Y, Baca S, Walsh B, Ferland D, Soubrier M, Choi HK, GrossW, Segal AM, Ludivico C, Puechal X.Amulticenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002;46:1309–1318.
18. Fava MP, Foradori GB, Garcia CB, Cruz FO, Aguilar JG, Kramer AS, Valdes FE. Percutaneoustransluminal angioplasty in patients with Takayasu arteritis: five-year experience.J Vasc Interv Radiol 1993;4:649–652.
19. 538. Fernandez Guerrero ML, Aguado JM, Arribas A, Lumbreras C, de Gorgolas M. The spectrum of cardiovascular infections due to Salmonella enterica: a reviewof clinical features and factors determining outcome. Medicine (Baltimore) 2004;83:123–138.
20. Both M, Aries PM, Muller-Hulsbeck S, Jahnke T, Schafer PJ, Gross WL, Heller M,
21. Reuter M. Balloon angioplasty of arteries of the upper extremities in patients with extracranial giant-cell arteritis. Ann Rheum Dis 2006;65:1124–1130.
22. Hunder GG, Bloch DA, Michel BA, Stevens MB, Arend WP, Calabrese LH, Edworthy SM, Fauci AS, Leavitt RY, Lie JT, Lightfoot RW Jr., Masi AT, McShane DJ, Mills JA,Wclassification of giant cell arteritis. Arthritis Rheum 1990;33:1122–1128.
23. Olin JW. Thromboangiitis obliterans (Buerger’s disease). N Engl J Med. 2000; 343:864–869.
24. Iwai T, Inoue Y, Umeda M, Huang Y, Kurihara N, Koike M, Ishikawa I. Oral bacteria in the occluded arteries of patients with Buerger disease. J Vasc Surg. 2005; 42: 107–115.
25. Adar R, Papa MZ, Halpern Z, Mozes M, Shoshan S, Sofer B, Zinger H, Dayan M, Mozes E. Cellular sensitivity to collagen in thromboangiitis obliterans. N Engl J Med. 1983; 308: 1113–1116.
26. Eichhorn J, Sima D, Lindschau C, Turowski A, Schmidt H, Schneider W, Haller H, Luft FC. Antiendothelial cell antibodies in thromboangiitis obliterans. Am J Med Sci. 1998; 315: 17–23.
27. Avcu F, Akar E, Demirkilic U, Yilmaz E, Akar N, Yalcin A. The role of prothrombotic mutations in patients with Buerger’s disease. Thromb Res. 2000; 100: 143–147.
28. Maslowski L, McBane R, Alexewicz P, Wysokinski WE. Antiphospholipid antibodies in thromboangiitis obliterans. Vasc Med. 2002; 7: 259–264.
29. Bozkurt AK, Koksal C, Ercan M. The altered hemorheologic parameters in thromboangiitis obliterans: a new insight. Clin Appl Thromb Hemost. 2004; 10: 45–50.
30. Ohta T, Ishioashi H, Hosaka M, Sugimoto I. Clinical and social consequences of Buerger disease. J Vasc Surg. 2004; 39: 176–180.
31. Cooper LT, Henderson SS, Ballman KV, Offord KP, Tse TS, Holmes DR, Hurt RD. A prospective, case-control study of tobacco dependence in thromboangiitis obliterans (Buerger’s disease). Angiology. 2006; 57: 73–78.
32. Sasajima T, Kubo Y, Inaba M, Goh K, Azuma N. Role of infrainguinal bypass in Buerger’s disease: an eighteen-year experience. Eur J Vasc Endovasc Surg. 1997; 13: 186–192.
33. Fiessinger JN, Schafer M. Trial of iloprost versus aspirin treatment for critical limb ischaemia of thromboangiitis obliterans: the TAO Study. Lancet. 1990; 335: 555–557.
34. Montori VM, Kavros SJ, Walsh EE, Rooke TW. Intermittent compression pump for nonhealing wounds in patients with limb ischemia: the Mayo Clinic experience (1998–2000). Int Angiol. 2002; 21: 360–366.
35. Donas KP, Schulte S, Ktenidis K, Horsch S. The role of epidural spinal cord stimulation in the treatment of Buerger’s disease. J Vasc Surg. 2005; 41: 830–836.
36. Isner JM, Baumgartner I, Rauh G, Schainfeld R, Blair R, Manor O, Razvi S, Symes JF. Treatment of thromboangiitis obliterans (Buerger’s disease) by intramuscular gene transfer of vascular endothelial growth factor: preliminary clinical results. J Vasc Surg. 1998; 28: 964–973.
37. Saito S, Nishikawa K, Obata H, Goto F. Autologous bone marrow transplantation and hyperbaric oxygen therapy for patients with thromboangiitis obliterans. Angiology. 2007; 58: 429–434.
38. Matoba S, Tatsumi T, Murohara T, Imaizumi T, Katsuda Y, Ito M, Saito Y, Uemura S, Suzuki H, Fukumoto S, Yamamoto Y, Onodera R, Teramukai S, Fukushima M, Matsubara H. Long-term clinical outcome after intramuscular implantation of bone marrow mononuclear cells (Therapeutic Angiogenesis by Cell Transplantation [TACT] trial) in patients with chronic limb ischemia. Am Heart J. 2008; 156: 1010–1018.
39. Durdu S, Akar AR, Arat M, Sancak T, Eren NT, Ozyurda U. Autologous bone-marrow mononuclear cell implantation for patients with Rutherford grade II-III thromboangiitis obliterans. J Vasc Surg. 2006; 44: 732–739.
40. Miyamoto K, Nishigami K, Nagaya N, Akutsu K, Chiku M, Kamei M, Soma T, Miyata S, Higashi M, Tanaka R, Nakatani T, Nonogi H, Takeshita S. Unblinded pilot study of autologous transplantation of bone marrow mononuclear cells in patients with thromboangiitis obliterans. Circulation. 2006; 114: 2679–2684.
41. Luscher TF, Keller HM, Imhof HG, et al. Fibromuscular hyper- plasia: extension of the disease and therapeutic outcome: results of the University Hospital Zurich Cooperative Study on Fibromuscular Hyperplasia. Nephron 1986;44 suppl 1:109-14.
42. Archibald GR, Beckmann CF, Libertino JA. Focal renal artery stenosis caused by fibromuscular dysplasia: treatment by percuta- neous transluminal angioplasty. AJR Am J Roentgenol 1988;151:593-6.
43. Cluzel P, Raynaud A, Beyssen B, et al. Stenoses of renal branch arteries in fibromuscular dysplasia: results of percutaneous trans- luminal angioplasty. Radiology 1994;193:227-32.
44. Mounier-Vehier C, Haulon S, Devos P, et al. Renal atrophy out- come after revascularization in fibromuscular dysplasia disease. J Endovasc Ther 2002;9:605-13.
45. Stanley JC, Gewertz BL, Bove EL, et al. Arterial fibrodysplasia: histopathologic character and current etiologic concepts. Arch Surg 1975;110:561-6.
46. Stanley JC, Wakefield TW. Arterial fibrodysplasia. In: Rutherford RB, ed. Vascular Surgery 6th ed. Philadelphia, Pa: Saunders; 2004:387-408.
47. Messina LM, Stanley JC. Renal artery fibrodysplasia and reno- vascular hypertension. In: Rutherford RB, ed. Vascular Surgery 6th ed. Philadelphia, Pa: Saunders; 2004:1650-64.
48. Mettinger KL. Fibromuscular dysplasia and the brain. II. Current concept of the disease. Stroke 1982;13:53-8.
49. Cloft HJ, Kallmes DF, Kallmes MH, et al. Prevalence of cerebral aneurysms in patients with fibromuscular dyspl