Capítulo 14 - Arteriopatías Vasospásticas
14.1 Acrocianosis
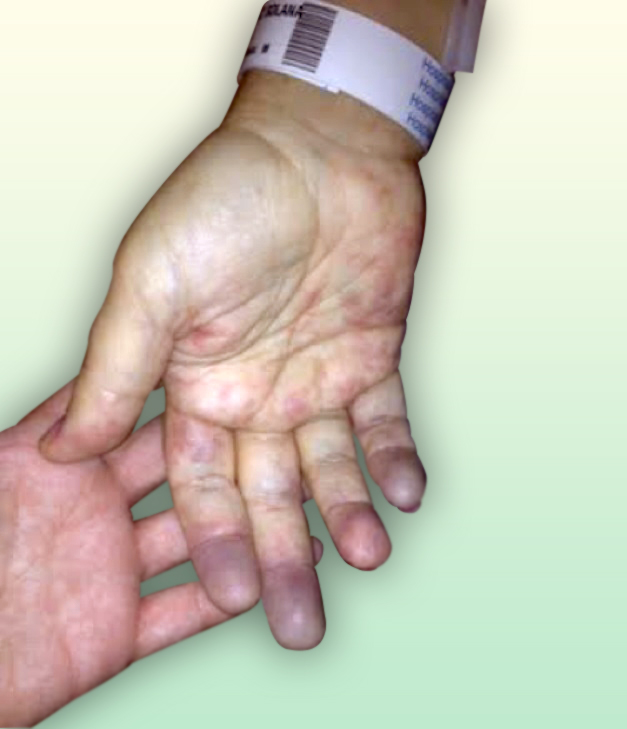
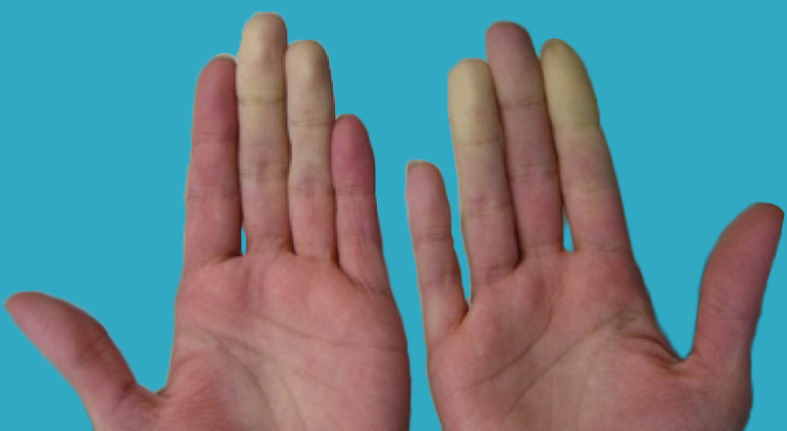
La acrocianosis es una cianosis simétrica, indolora y persistente de las manos, y con menor frecuencia de los pies o de la cara, causada por el vasospasmo persistente de los pequeños vasos de la piel en respuesta al frío.
La acrocianosis primaria no tiene una causa identificable. En su forma secundaria puede relacionarse con enfermedades del colágeno, trastornos hematológicos, trastornos neurovasculares, fármacos, toxinas, infecciones, metabolopatías hereditarias y algunas neoplasias malignas.
Se ha propuesto varios posibles mecanismos fisiopatológicos como: anomalía del tono de las arteriolas, actividad anormal del sistema simpático o alteración de la respuesta microvascular con dilatación y estasis de capilares y vénulas. Se ha podido observar que la acrocianosis se produce por una vasoconstricción arterial y del esfínter precapilar asociado a dilatación de capilares y del plexo venoso subdérmico. Se cree que la vasoconstricción arterial se produce por hipersensibilidad al frío y la dilatación post capilar por respuesta exagerada del sistema nervioso simpático.
La acrocianosis primaria es más frecuente mujeres (6:1), con menos de 30 años y no se asocia con enfermedad arterial oclusiva. Suelen estar asintomáticos salvo por la cianosis, los pulsos son normales y la palma de las manos están húmedas. A diferencia del Raynaud la cianosis es persistente y no episódica, no existe fase de palidez, no se producen cambios tróficos ni úlceras, suele ser simétrica y el paciente no presenta dolor. Este cuadro llega a estar presente en el 20% de las pacientes con anorexia nerviosa.
El diagnóstico en la acrocianosis primaria suele basarse en la historia clínica y exploración física. La única prueba en la que se detectan posibles alteraciones es en la capilaroscopia pero no hay signos patognomónicos. En la acrocianosis secundaria nos debe orientar los antecedentes del pacientes, alteraciones en la exploración física (ausencia de pulsos, lesiones cutáneas,
ingesta de fármacos,…) y es preciso estudios analíticos para descartar enfermedades hematológicas o autoinmunes.
El diagnóstico diferencial debe realizarse con el síndrome de Raynaud, livedo reticularis y eritema pernio.
Además de tranquilizar al paciente y evitar el frío, no suele requerir tratamiento. Puede intentarse la administración de vasodilatadores, aunque en general son ineficaces.
La acrocianosis secundaria puede ser consecuencia de hipoxemia, enfermedades de tejido conectivo, ateroembolia, anticuerpos antifosfolípidos, crioaglutininas o crioglobulinas y acompaña a la anorexia nerviosa y al síndrome de taquicardia ortostática. El tratamiento se orientaráa la patología basal.
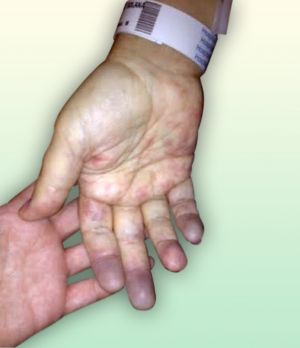
Fig. 1 Acrocianosis en manos
14.2 Fenómeno de Raynaud
14.2.1 Definición
El Síndrome de Raynaud es un trastorno que se caracteriza por la disminución episódica del flujo sanguíneo - generalmente en los dedos de las manos, y con menor frecuencia, en las orejas, los dedos de los pies, los pezones, las rodillas o la nariz - que se manifiesta con palidez o cianosis producido por vasoconstricción de las pequeñas arterias o arteriolas de los dedos.
En general, los espasmos vasculares se originan como ataques en respuesta a la exposición al frío, vibraciones o a una alteración emocional.
El Síndrome de Raynaud puede ocurrir como una única manifestación (Enfermedad de Raynaud o Síndrome de Raynaud primario) sin enfermedad subyacente o secundario a otras enfermedades (Fenómeno de Raynaud o Síndrome de Raynaud secundario).
Las enfermedades que se asocian más frecuentemente con el Fenómeno de Raynaud son las autoinmunes, las del tejido conectivo y aquellas que tienen efecto vasospástico tales como:
Conectivopatias: Lupus eritematoso sistémico. La esclerodermia. El síndrome CREST (depósitos de calcio en la piel, fenómeno de Raynaud, dismotilidad esofágica, esclerodactilia, telangiectasia). La artritis reumatoide. La polimiositis. Dermatomiositis. Síndrome de Sjögren.
Enfermedad vascular oclusiva: Aterosclerosis. La enfermedad de Buerger. Síndrome salida de tórax.
Hipertensión pulmonar.
Alteraciones neurológicas: Discopatías. Siringomelia. Tumores medulares. Ictus. Poliomielitis. Síndrome del túnel del carpo.
Discrasias sanguíneas: Crioglobulinemia. Crioaglutininas. Criofibrinogenemia. Macroglobulinemia de Waldenström.
Traumatismos: Lesiones por vibraciones (Síndrome del martillo, de la mecanógrafa, del pianista,..). Descargas eléctricas.
Fármacos: Derivados ergotamínicos. Metisergida. Bloqueantes beta adrenérgicos. Bleomicina. Vinblastina. Cisplatino.
14.2.2 Epidemiología
Afecta en España entre el 3’7% y el 5% de toda la población y generalmente de una forma leve pero en el 10% de los casos adopta formas de presentación severas. Es mas frecuente entre mujeres que entre hombres (en los primeros estudios 4:1 pero en valoraciones recientes la proporción es de 1:1,6) y especialmente entre los veinte y cuarenta años aunque puede aparecer a cualquier edad, incluso en niños.
También aparece con cierta frecuencia en la menopausia, especialmente entre aquellas mujeres que no han realizado tratamiento hormonal sustitutivo.
En dos tercios de los casos la sintomatología desaparece espontáneamente en los 7 años siguientes a su aparición.
14.2.3 Etiología
Se desconoce cual es la causa exacta. Originalmente, Raynaud propuso que la isquemia digital episódica inducida por el frío era secundaria a una intensa vasoconstricción refleja simpática. Esta teoría es apoyada por el hecho de que los bloqueadores alfa y la simpatectomía disminuyen la frecuencia y gravedad del fenómeno en algunos pacientes. Una hipótesis alternativa es que existe un aumento de la respuesta al frío o a los estímulos simpáticos normales. También es posible que la vasoconstricción simpática refleja normal se superponga a una enfermedad vascular digital local o que exista aumento de la actividad adrenérgica neuroefectora.
14.2.4 Factores de Riesgo
Hay ciertas enfermedades o elecciones de estilo de vida que pueden aumentar el riesgo de que una persona desarrolle el fenómeno de Raynaud. Los factores de riesgo son los siguientes:
- Una enfermedad autoinmune o del tejido conectivo existente.
- El hábito de fumar (en los hombres).
- El consumo de alcohol (en las mujeres).
- El Helicobacter pylori (H. pylori)
14.2.5 Clínica
Los síntomas más comunes del fenómeno de Raynaud pueden incluir:
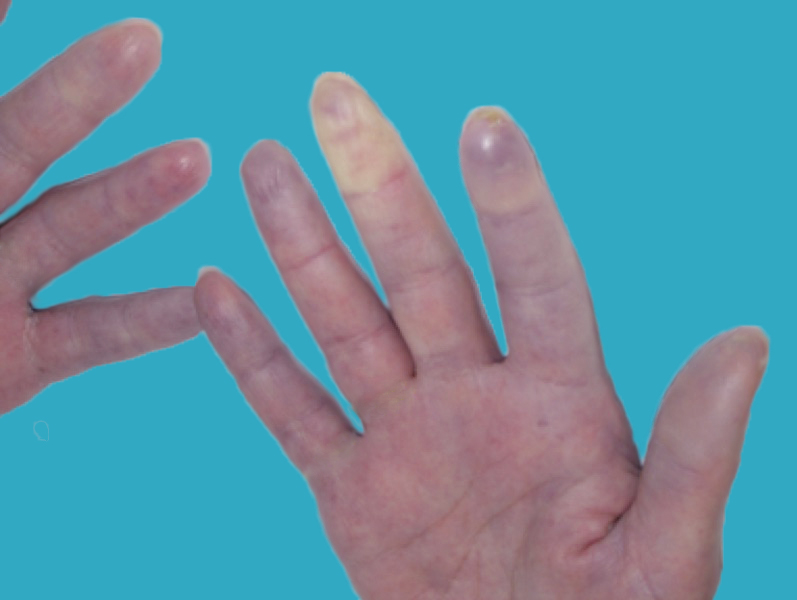
- Un patrón de cambios de color en los dedos de la siguiente forma: Pálido o blanco que se torna azul y luego rojo cuando las manos se calientan; los cambios de color generalmente ocurren tras la exposición al frío o tras una alteración emocional.
- Las manos pueden hincharse o doler cuando se calientan.
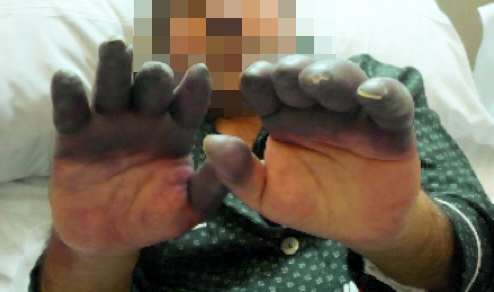
- En los casos graves se desarrollan úlceras en los pulpejos de los dedos.
- Se puede desarrollar gangrena en los dedos, lo que puede llevar a una amputación (en alrededor del 10 por ciento de los casos graves).
- En una cuarta parte de los pacientes puede asociarse a anginas de Prinzmetal y jaquecas migrañosas.
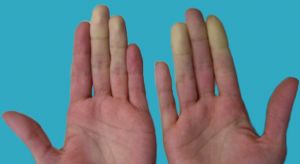
Fig. 2 Fase de palidez
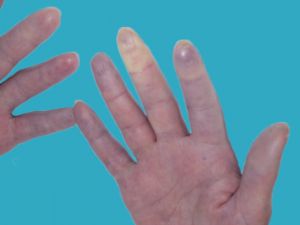
Fig. 3 Fase cianosis
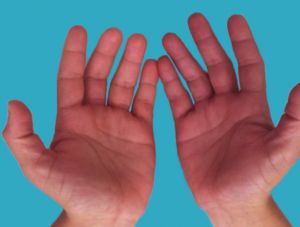
Fig. 4 Fase eritematosa
14.2.6 Diagnóstico
Principalmente por la clínica que cuenta el paciente: crisis episódicas de palidez o cianosis bien delimitadas de uno o más dedos producida por la exposición al frío o las emociones.
No hay exámenes de laboratorio específicos que puedan confirmar el diagnóstico del fenómeno de Raynaud. Por eso el diagnóstico suele basarse en los síntomas que describe el paciente y en la exclusión de otras enfermedades.
En casos asociados a Hipertensión Pulmonar puede auscultarse un aumento del ruido de cierre de la válvula pulmonar. La exploración arterial debe incluir todos los vasos de la extremidad y suele ser normal. El test de Allen debe realizarse en todo los pacientes para descartar oclusión de la arteria cubital. La inspección detallada de la piel puede poner en evidencia telangiectasias en dedos o esclerodactilia con engrosamiento de la piel. Los hematomas subungueales son frecuentes en microtraumatismos de repetición en manos pero también en microembolismos de colesterol. Hay que buscar posibles ulceraciones de dedos, fóveas, pigmentaciones moteadas, infartos del borde ungueal o piel tensa y brillante que hagan sospechar conectivopatias.
Se pueden realizar pruebas de provocación con frío para ver los cambios de color en las manos y estudiar los cambios de curva del flujo digital con un pletismógrafo o estudios de capilaroscopia subungueal. Estos cambios son más intensos en pacientes con Síndrome de Raynaud que en personas sanos. Estudios de flujo, Doppler láser termografía,…son estudios más académicos que útiles en la práctica.
Los estudios de imagen con AngioRNM o arteriografía hay que dejarlos para casos severos.
14.2.7 Tratamiento
El tratamiento individualizado del Síndrome de Raynaud se basará:
- Edad, estado general de salud y antecedentes médicos.
- Lafrecuencia, intensidad y riesgo de desarrollar gangrena tiene la sintomatología de la enfermedad.
- Tolerancia a determinados medicamentos, procedimientos o terapias.
- Expectativas para la evolución de la enfermedad
- Opinión o preferencia.
Aunque no hay cura para el fenómeno de Raynaud, el trastorno puede controlarse exitosamente con un tratamiento adecuado. Se pueden dividir en tres grupos: terapias conductuales, tratamiento farmacológico y procedimientos intervencionistas/quirúrgicos.
Las terapias conductuales están indicadas en todos los paciente (y las la única necesaria en pacientes con síntomas leves) incluyen lo siguiente:
- Medidas preventivas tales como el uso de guantes o evitar la exposición al frío. Tener en cuenta el concepto de “calor corporal total”
- Dejar de fumar ( la nicotina es un potente vasoconstrictor ).
- Usar protectores para los dedos ulcerados.
- Evitar traumatismos o vibración en la mano (tales como el uso de herramientas vibratorias).
Los tratamientos farmacológicos están indicados en pacientes con sintomatología grave. En síntomas moderados solo se requieren en épocas invernales. Solo entre el 50 y 75% de los pacientes consiguen mejoría con fármacos:
Los antagonistas del calcio son los fármacos más utilizados. Actúan bloqueando los canales del calcio de la membrana celular del músculo liso y provocando su relajación y por tanto vasodilatación directa. Existen tres grupos: dihidropiridinas (Nifedipino 30-60 mg diarios), son los más potentes pero como efecto secundarios pueden producir hipotensión, sofocos y edema; Diltiazem, menos potente y con menos efectos secundarios, y Verapamilo, es el menos eficaz. Son útiles en el Raynaud primario pero con escaso efecto en el Raynaud secundario.
Los Bloqueantes alfa adrenérgicos. Son la Prasozina (acción corta), Terazosina y Doxazosina (acción larga). Actúan inhibiendo los receptores alfa1 post sinápticos bloqueando la acción de la noradrenalina sobre el músculo liso vascular. El Losartan (50- mg día ) es un antagonista del receptor II de tipo 1 y ha demostrado mejores resultados que el nifedipino. Son útiles tanto en el Raynaud primario como en el Raynaud secundario
Los antiagregantes no han demostrado ser útiles como tratamiento.
La Fluoxetina es un inhibidor específico de la recaptación de serotonina y a dosis de 20 mg diario ha demostrado disminuir significativamente la frecuencia de vasospasmos en el Raynaud.
Los inhibidores de la endotelina como el Bosentán pueden bloquear la vasoconstricción y mejorar los procesos vasospásticos.
Las Prostaglandinas E1/Prostaciclinas. Son vasodilatadores potentes de uso IV que se utilizan en casos de isquemia crítica.La PGE1se administra a dosis IV o IA de ng/kg/min durante 72 horas, tiene un efecto potente pero de corta duración y su principal uso es la curación de úlceras. El Iloprost es una análogo sintético de la prostaciclina que disminuye la intensidad y frecuencia de las crisis de Raynaud. Se administra a dosis de 2 ng/kg/min durante 8 horas y durante 5 días y posteriormente una vez cada 6 semanas.
Los procedimientos no farmacológicos de tipo intervencionista o quirúrgico incluyen: la terapia de bomba de compresión neumática, los estimuladores de la médula espinal, la simpatectomía cervicotorácica, las reconstrucciones vasculares para vasos obstruidos y amputaciones.

Fig. 5 Necrosis dedos en paciente con S. de Raynaud
14.3 Eritromelagia
14.3.1 Concepto
Eritromelalgia es una patología infrecuente caracterizada por la tríada de: eritema, calor y dolor en extremidades.
14.3.2 Incidencia y Prevalencia
La incidencia y prevalencia de esta patología son complicadas de establecer debido a la falta de estudios y la rareza de la enfermedad. Se sospecha que es más frecuente de lo que aparece en los estudios, ya que dada la escasa gravedad, no es considerado por los pacientes como motivo de consulta. Un estudio en la población de Olmsted (2009) aportaba los siguientes resultados: incidencia: 1,3 casos por cien mil de habitantes y año (primaria 1,1 y secundaria 0,2) y mostraba que los casos habían aumentado en los últimos 30 años al compararlos con el estudio poblacional realizado en Noruega donde la incidencia era cinco meces menor.
14.3.3 Etiología y Clasificación
La etiología de eritromelalgia es desconocida.
Clasificaremos dos grandes grupos:
Eritermalgia primaria. Incluiría todos los casos en que desconocemos la causa desencadenante Algunos estudios lo han asociado a desorden congénito relacionado con mutaciones en el gen SCN9A que codifica funcionalmente los canales de sodio Nav 1.7.
Eritermalgia secundaria. Se incluirían todos aquellos casos asociados a trombocitemia en síndromes mieloproliferativos, sobre todo trombocitosis esencial y policitemia vera. También como efecto secundario de algunas drogas: vasoactivas, antagonistas del calcio; o asociado a otras patologías subyacentes: gota, LES, artritis reumatoide, crioglobulinemia, endarteritis obliterante, tromboangeitis obliterante, poliarteritis nodosa, arteriosclerosis, diabetes mellitus, enfermedades neurológicas y vasculares.
14.3.4 Fisiopatología
La fisiopatología de este síndrome es desconocida, pero existen diversas hipótesis:
La hipótesis vascular constata un doble hallazgo contradictorio, por un lado existe aumento de flujo vascular local, por otro lado se acompaña de hipoxia en la región. Este doble efecto se produciría por cortocircuitos de fístulas arteriovenosas a este nivel, que se permeabilizan con el aumento de temperatura, y que provocarían hipoxemia por bajo flujo en plexos capilares mas superficiales y aumento global del flujo en la piel.
La hipótesis neurológica encuentra una asociación importante entre eritromelalgia y neuropatías periféricas. Esta asociación se expresaría a través de alteraciones de los receptores de las fibras C en pieles sensibilizadas, que se activarían cuando el calor alcanza los 32-36ºC y desencadenan vasodilatación mediante reflejos axónicos.
14.3.5 Clínica y Diagnóstico
El diagnóstico se basa en la historia clínica y en la exploración física, sin necesidad de exploraciones complementarias. El cuadro clínico típico es la elevación de la temperatura cutánea asociado a eritema e intenso dolor, más frecuentemente de afectación simétrica y de miembros inferiores.El dolor es intermitente, con periodos de tiempo de mayor dolor denominados “ataques”, que varían desde minutos a horas; la duración y gravedad de los síntomas aumenta en verano y pueden ser inducidas por el ejercicio físico, descendiendo o presionando la extremidad afecta. Algunos pacientes refieren empeoramiento con la ingesta de alcohol. Por otro lado, refieren mejoría con el enfriamiento o con la elevación de la extremidad afecta.
La biopsia cutánea demuestra en algunas series de pacientes trombos intravasculares y proliferación de la íntima que no se han demostrado en otras.
Las pruebas complementarias en este síndrome van encaminadas a diagnosticar procesos subyacentes en la eritromelalgia secundaria. Se aconseja recuento y fórmula de la serie roja y blanca, bioquímica sérica con determinación de glucosa sérica y anticuerpos antinucleares (Mork, 2000). En los casos de historia familiar de eritromelalgia se debe pedir estudio genético para buscar mutaciones del SCN9A.
14.3.6 Diagnóstico Diferencial
La eritromelalgia es un cuadro clínico cuyo diagnóstico, según se ha expuesto anteriormente, se realiza con la historia clínica y la exploración física. Pero en un estadio precoz de la enfermedad, pueden no estar presentes todos los síntomas y signos, y debemos realizar el diagnóstico diferencial con una serie de patologías.

14.3.7 Pronóstico y Complicaciones
El pronóstico de la enfermedad depende del tipo, de la gravedad de los síntomas y de la respuesta al tratamiento. En general tiene una esperanza de vida sensiblemente menor que el resto de la población.
Pocos pacientes curan completamente, el resto un tercio empeora progresivamente, otro se estabiliza y el tercero suelo mejorar. La calidad de vida de estos pacientes es mala, especialmente en el apartado de funcionamiento físico.
la simpatectomía cervicotorácica, las reconstrucciones vasculares para vasos obstruidos y amputaciones.
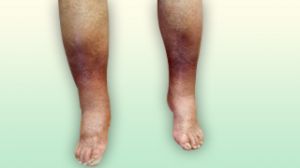
Fig. 6 Eritromelalgia
Los pacientes pueden presentar úlceras dérmicas y múltiples lesiones cutáneas secundarias a los métodos de enfriamiento de la extremidad afecta para aliviar el dolor. En estadios avanzados podemos encontrar lesiones graves de isquemia e incluso gangrena.
14.3.8 Tratamiento
En la eritromelalgia secundaria a cuadros mieloproliferativos, el tratamiento con ácido acetilsalicílico a bajas dosis (40-100 mg) o indometacina, junto al tratamiento de la trombocitosis, han demostrado ser efectivos en el control de la enfermedad. Las flebotomías en la policitemia vera no han demostrado eficacia. Si el proceso mieloproliferativo cursa con historia de episodios de sangrado, el ácido acetilsalicílico estácontraindicado.
En la eritromelalgia secundaria, el tratamiento y/o control de la patología subyacente mejora el cuadro clinico.
En la eritromelalgia primaria, el tratamiento es el del dolor neuropático con analgésicos tópicos, orales, opioides, anticomiciales, antidepresivos, etc. También se ha utilizado lidocaína y ketamina endovenosa y parches de lidocaína. La respuesta de los pacientes a estos tratamientos es muy variable y su eficacia, controvertida.
El tratamiento de las complicaciones, como las dermatitis o las úlceras vasculares, se realizaría con medicación tópica y curas locales a cargo de enfermería.
A continuación se muestra una tabla con el esquema de las opciones de tratamiento más importantes.

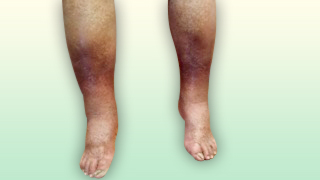
14.4 Livedo Reticularis
En este proceso, zonas localizadas de las extremidades adquieren un aspecto moteado o reticular de color rojizo o azulado. El aspecto moteado puede ser más evidente tras la exposición al frío.
Se conocen las formas primaria y secundaria
de la livedo reticular.
La primaria o idiopática puede ser benigna o acompañarse de úlceras. La forma benigna surge con mayor frecuencia en mujeres que en varones y el tercer decenio es el lapso más frecuente de comienzo.
Los individuos con la forma benigna por lo común no tienen síntomas y buscan la atención médica por razones estéticas. Habráque tranquilizarlos verbalmente y recomendarles que no se expongan a entornos fríos. No conviene farmacoterapia alguna.
La livedo reticularis primaria con úlceras también recibe el nombre de atrofia blanca en placas. Las úlceras son dolorosas y tardan meses para curar.
La livedo secundaria puede surgir con ateroembolia, lupus eritematoso generalizado y otras vasculitis, anticuerpos contra cardiolipina, hiperviscosidad, crioglobulinemia y síndrome de Sendín (accidente isquémico livedo reticular). En raras ocasiones surgen úlceras de la piel.
la simpatectomía cervicotorácica, las reconstrucciones vasculares para vasos obstruidos y amputaciones.
Fig. 7 Livedo Reticularis en piernas
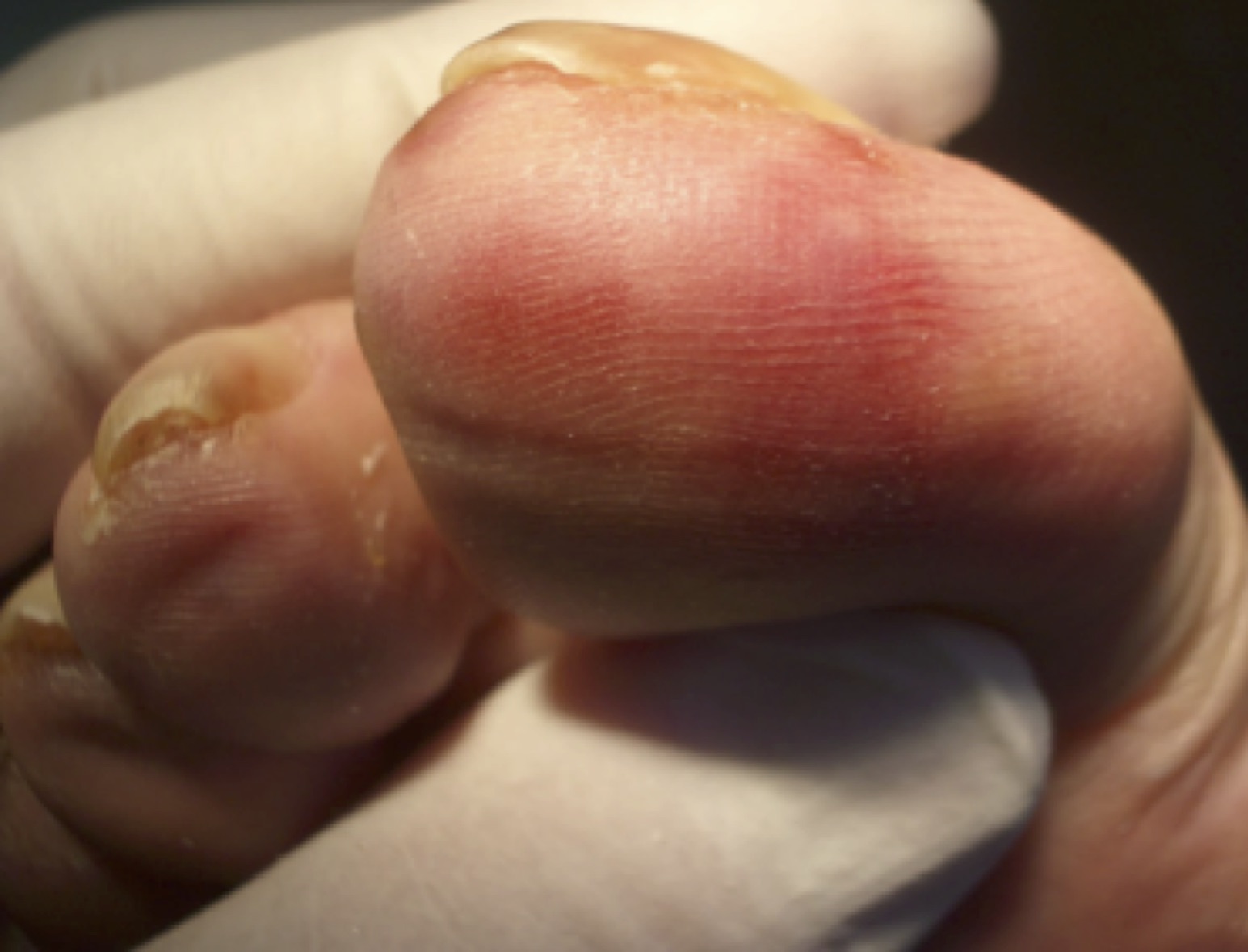
14.5 Eritema Pernio (Sabañones)
Se trata de un trastorno vasculítico asociado a la exposición al frío. A bajas temperaturas se desarrollan lesiones elevadas eritematosas en la parte baja de las piernas y en los pies, siendo características las lesiones en el dorso de las falanges proximales. Éstas cursan con prurito y sensación urente y pueden evolucionar a ampollas y úlceras.
Su frecuencia ha disminuido en los últimos años. Se asocia fundamentalmente a climas fríos y húmedos.
En la mayoría de los casos las lesiones se asocian a la exposición de frío y humedad pero también se presentan de forma secundaria en cuadros de elevación sanguínea de crioproteínas, conectivopatías y leucemia, especialmente en casos de lesiones de larga duración, mas alláde la exposición al frío.
La fisiopatología es similar al Raynaud con vasoconstricción arterial, dilatación de vénulas post capilares y aumento de la viscosidad sanguínea tras exposición al frío. Puede coexistir clínicamente con el síndrome de Raynaud pero no se conoce por que se desarrollan en unos casos eritema pernio y en otros Raynaud.
El estudio anatomopatológico muestra una vasculitis caracterizada por proliferación de la íntima e infiltración perivascular constituida por mononucleares y polimorfonucleares. En el tejido subcutáneo puede haber células gigantes.
Suele presentarse en mujeres adolescentes pero puede aparecer en cualquier sexo y edad. Las lesiones aparecen en dedos de pies o manos, en el dorso, en los meses de frío, y pueden variar en número y tamaño. Presentan dolor, quemazón o prurito. Al tacto las lesiones pueden notarse mas frías que los tejidos que las rodean. En los casos agudos pueden autolimitarse y desaparecer en unos días.
El diagnóstico es clínico, basado en la anamnesis y la exploración física. En los eritemas pernios primarios el resto de estudios diagnósticos es normal pero es importante descatar los secundarios a enfermedad autoinmune realizando una analítica con hemograma, fórmula y recuento, ANA, factor reumatoide, crioglobulinas, criofibrinógeno, crioaglutininas y viscosidad plasmática.
El diagnóstico diferencial debe realizarse con microateroembolismos (a veces es preciso biopsia de la lesión), eritema indurado (enfermedad de Bazin), vasculitis nodular, eritema nodoso y paniculitis fría.
Los pacientes deben evitar la exposición al frío y las úlceras se deben mantener limpias y protegidas con a pósitos estériles. En algunos pacientes pueden ser útiles los antagonistas del calcio dihidropiridínicos (nifedipino 20 mg/8 horas de 7 a 10 días en la fase aguda) como alternativa se puede utilizar alfa-bloqueantes adrenérgicos (prazosina).
la simpatectomía cervicotorácica, las reconstrucciones vasculares para vasos obstruidos y amputaciones.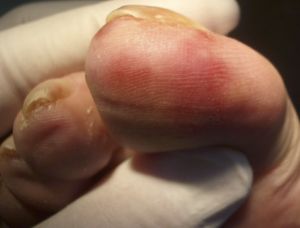
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fig. 8 Sabañones
14.6 Congelación
En este trastorno se produce una lesión hística por exposición al frío ambiental intenso o por contacto directo con un objeto muy frío. La lesión hística se debe tanto a la congelación como a la vasoconstricción.
La congelación afecta habitualmente a las zonas distales de las extremidades o partes expuestas de la cara, como las orejas, nariz, mentón o mejillas. La congelación superficial afecta a la piel y tejido subcutáneo. Los pacientes experimentan dolor o parestesias, y la piel aparece blanca y cérea. Tras el recalentamiento se producen cianosis y eritema, formación de ronchas, edema y vesículas superficiales. Las congelaciones profundas afectan al músculo, nervios y vasos sanguíneos más profundos. Esto puede dar lugar a edema de la mano o pie, vesículas y ampollas, necrosis hística y gangrena.
El tratamiento inicial es el recalentamiento, realizado en un medio en el que no se produzca una reexposición a condiciones de congelación. El recalentamiento se logra por inmersión de la parte afectada en un baño de agua a temperaturas de 40 a 44°C. El masaje, aplicación de agua helada y calor extremo están contraindicados. La zona herida debe limpiarse con jabón o antiséptico y se deben aplicar apósitos estériles. Durante el recalentamiento con frecuencia son necesarios los analgésicos. Si se observan signos de infección se administrarán antibióticos. No se ha establecido la eficacia de los fármacos bloqueadores simpáticos. Tras la recuperación, la extremidad afectada puede mostrar mayor sensibilidad al frío.
14.7 Bibliografía
1. Medicina Vascular. Complemento de Braunwald. Tratado de Cardiología. 2ª Edición. Mark A. Greager, Joshua A. Beckman. Edt Elservier Saunders 2013.
2. Rutherford´s Vascular Surgery. 8Th Edition. Rutherford. Jack L. Cronenwett MD, K. Wayne Jhonston. Elservier Saunders 2014.
3. M.W. Neumeister: Botulinum toxin type A in the treatment of Raynaud’s phenomenon. J Hand Surg Am. 35:2085-2092 2010
4. Fregene, D. Ditmars, A. Siddiqui: Botulinum toxin type A: a treatment option for digital ischemia in patients with Raynaud’s phenomenon. J Hand Surg Am. 34:446-452 2009
5. A.M. Yee, R.N. Hotchkiss, S.A. Paget: Adventitial stripping: a digit saving procedure in refractory Raynaud’s phenomenon. J Rheumatol. 25:269-276 1998
6. J.C. de Trafford, K. Lafferty, C.E. Potter, et al.: An epidemiological survey of Raynaud’s phenomenon. Eur J Vasc Surg. 2:167-170 1988
7. P. Sambo, D. Amico, R. Giacomelli, et al.: Intravenous N-acetylcysteine for treatment of Raynaud’s phenomenon secondary to systemic sclerosis: a pilot study. J Rheumatol. 28:2257-2262 2001
8. L. Chung, L. Shapiro, D. Fiorentino, et al.: MQX-503, a novel formulation of nitroglycerin, improves the severity of Raynaud’s phenomenon: a randomized, controlled trial. Arthritis Rheum. 60:870-877 2009
9. Abou-Raya, S. Abou-Raya, M. Helmii: Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol. 35:1801-1808 2008
10. F.M. Wigley: Clinical practice. Raynaud’s Phenomenon. N Engl J Med. 347:1001-1008 2002
11. A.E. Smyth, A.L. Bell, I.N. Bruce, et al.: Digital vascular responses and serum endothelin-1 concentrations in primary and secondary Raynaud’s phenomenon. Ann Rheum Dis. 59:870-874 2000
12. V.A. Nguyen, K. Eisendle, I. Gruber, et al.: Effect of the dual endothelin receptor antagonist bosentan on Raynaud’s phenomenon secondary to systemic sclerosis: a double-blind prospective, randomized, placebo-controlled pilot study. Rheumatology (Oxford). 49:583-587 2010
13. M. Matucci-Cerinic, C.P. Denton, D.E. Furst, et al.: Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 70:32-38 2011
14. D. Aletaha, T. Neogi, A.J. Silman, et al.: 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 62:2569-2581 2010
15. M. Garcia-Carrasco, A. Siso, M. Ramos-Casals, et al.: Raynaud’s phenomenon in primary Sjogren’s syndrome. Prevalence and clinical characteristics in a series of 320 patients. J Rheumatol.29:726-730 2002
16. H. Nakamura, A. Kawakami, T. Hayashi, et al.: Anti-centromere antibody-seropositive Sjogren’s syndrome differs from conventional subgroup in clinical and pathological study. BMC Musculoskelet Disord. 11:140 2010
17. S.N. Lambova, S.I. Kuzmanova: Raynaud’s phenomenon in common rheumatic diseases. Folia Med (Plovdiv). 48:22-28 2006
18. O.Trejo, M. Ramos-Casals, M. Garcia-Carrasco, et al.: Cryoglobulinemia: study of etiologic factors and clinical and immunologic features in 443 patients from a single center. Medicine (Baltimore). 80:252-262 2001
19. C.G. Kuhar, T. Mesti, B. Zakotnik: Digital ischemic events related to gemcitabine: report of two cases and a systematic review. Radiol Oncol. 44:257-261 2010
20. E.K. Schildmann, A.N. Davies: Paraneoplastic Raynaud’s phenomenon–good palliation after a multidisciplinary approach. J Pain Symptom Manage. 39:779-783 2010
21. S. Lambova, U. Muller-Ladner: Capillaroscopic pattern in paraneoplastic Raynaud’s phenomenon. Rheumatol Int. 2011 published online 21 Jan
22. J.D. Coffman: Raynaud’s phenomenon. Curr Treat Options Cardiovasc Med. 2:219-226 2000
23. B. Coleiro, S.E. Marshall, C.P. Denton, et al.: Treatment of Raynaud’s phenomenon with the selective serotonin reuptake inhibitor fluoxetine. Rheumatology (Oxford). 40:1038-1043 2001
24. J. Rey, E. Cretel, R. Jean, et al.: Serotonin reuptake inhibitors, Raynaud’s phenomenon and erythromelalgia. Rheumatology (Oxford). 42:601-602 2003
25. N. Selenko-Gebauer, N. Duschek, G. Minimair, et al.: Successful treatment of patients with severe secondary Raynaud’s phenomenon with the endothelin receptor antagonist bosentan. Rheumatology (Oxford). 45 (Suppl 3):iii45-iii48 2006
26. M. Ramos-Casals, P. Brito-Zeron, N. Nardi, et al.: Successful treatment of severe Raynaud’s phenomenon with bosentan in four patients with systemic sclerosis. Rheumatology (Oxford). 43:1454-1456 2004
27. N. Tsifetaki, V. Botzoris, Y. Alamanos, et al.: Bosentan for digital ulcers in patients with systemic sclerosis: a prospective 3-year followup study. J Rheumatol. 36:1550-1552 2009
28. T.L. Levien: Phosphodiesterase inhibitors in Raynaud’s phenomenon. Ann Pharmacother. 40:1388-1393 2006
29. R. Fries, K. Shariat, H. von Wilmowsky, et al.: Sildenafil in the treatment of Raynaud’s phenomenon resistant to vasodilatory therapy. Circulation. 112:2980-2985 2005
30. P.D. Shenoy, S. Kumar, L.K. Jha, et al.: Efficacy of tadalafil in secondary Raynaud’s phenomenon resistant to vasodilator therapy: a double-blind randomized cross-over trial. Rheumatology (Oxford). 49:2420-2428 2010
31. A.L. Herrick, F. van den Hoogen, A. Gabrielli, et al.: Modified-release sildenafil reduces Raynaud’s phenomenon attack frequency in limited cutaneous systemic sclerosis. Arthritis Rheum. 63:775-782 2011
32. J.D. Coffman: Raynaud phenomenon. 1989 Oxford University Press New York
33. P.H. Carpentier: Definition and epidemiology of vascular acrosyndromes. Rev Prat. 48:1641-1646 1998
34. E. Davis: Clinical aspects of acrocyanosis. Adv Microcirc. 10:101-106 1982
35. M.A. Creager, V.J. Dzau: Vascular disease of the extremity. D.L. Kasper A.S. Fauci D.L. Lango Harrison’s principles of internal medicine. ed 16 2005 McGraw-Hill New York 1490-1495
36. C. Hediger, B. Rost, P. Itin: Cutaneous manifestations in anorexia nervosa. Schweiz Med Wochenschr. 130:565-575 2000
37. P.J. Brown, M.J. Zirwas, J.C. English: The purple digit an algorithmic approach to diagnosis. Am J Clin Dermatol. 11:103-116 2010
38. Mansilha, S. Sampaio: Vasospastic disorders of the upper extremities. C.D. Liapis K. Balzer F. Benedetti-Valentini J. Fernandes European manual of medicine: Vascular surgery. 2007 Springer-Verlag Berlin 237-246 Part 3
39. A.K. Kurklinsky, V.M. Miller, T.W. Rooke: Acrocyanosis: the Flying Dutchman. Vasc Med. 16:288-301 2011
40. R.A. Mangiafico, L.S. Malatino, M. Santonocito, et al.: Plasma endothelin-1 concentrations during cold exposure in essential acrocyanosis. Angiology. 47:1033-1038 1996
41. H. Heidrich: Functional vascular diseases: Raynaud’s syndrome, acrocyanosis and erythromelalgia. Vasa. 39:33-41 2010
42. G. Monticone, L. Colonna, G. Palmeri, et al.: Quantitative nailfold capillary microscopy findings in patients with acrocyanosis compared with patients having systemic sclerosis and normal subjects. J Am Acad Dermatol. 42:787-790 2000
43. H.C. Nousari, A. Kimyai-Asadi, G.J. Anhalt: Chronic idiopathic acrocyanosis. J Am Acad Dermatol. 45:S207-S208 2001
44. S.G. Golombek, S. Ally, P.K. Woolf: A newborn with cardiac failure secondary to a large vein of Galen malformation. South Med J. 97:516-518 2004
45. M.A. Jimenez, S. Polena, N.L. Coplan, et al.: Methemoglobinemia and transesophageal echo. Proc West Pharmacol Soc. 50:134-135 2007
46. E. Cholongitas, D. Ioannidou: Acrocyanosis due to cold agglutinins in a patient with rheumatoid arthritis. J Clin Rheumatol. 15:375 2009
47. Sinha, G. Richardson, R.T Patel: Cold agglutinin related acrocyanosis and paroxysmal haemolysis. Eur J Vasc Endovasc Surg. 30:563-565 2005
48. Y. Solak, S. Aksoy, S. Kilickap, et al.: Acrocyanosis as a presenting symptom of Hodgkin lymphoma. Am J Hematol. 81:151-152 2006
49. O. Trejo, M. Ramos-Casals, M. Garcia-Carrasco, et al.: Cryoglobulinemia: study of etiological factors and clinical and immunologic features in 443 patients from a single center. Medicine. 80:252-262 2001
50. R. Strumia: Skin signs in anorexia nervosa. Dermatoendocrinol. 1:268-270 2009
51. J.G. Richter, O. Sander, M. Schneider, et al.: Diagnostic algorithm for Raynaud’s phenomenon and vascular skin lesions in systemic lupus erythematosus. Lupus. 19:1087-1095 2010
52. M.J. Diógenes, P.C. Diógenes, R.M. de Morais Carneiro, et al.: Cutaneous manifestations associated with antiphospholipid antibodies. Int J Dermatol. 43:632-637 2004
53. R. Ozaras, M. Yemisen, B. Mete, et al.: Acrocyancosis developed with amphotericin B deoxycholate but not amphotericin B lipid complex. Mycoses. 50:242 2007
54. A.H. Hall: Chronic arsenic poisoning. Toxicol Lett. 128:69-72 2002
55. E. Glazer, J.P. Pacanowski, L.R. Leon: Asymptomatic lower extremity acrocyanosis: report of two cases and review of the literature. Vascular. 19:105-110 2011
56. J.M. Steward, M.H. Gewitz, A. Weldon, et al.: Patterns of orthostatic intolerance: the orthostatic tachycardia syndrome and adolescent chronic fatigue. J Pediatr. 135:218-221 1999
57. C. Boderman, A. Rötig, P. Rustin, et al.: Hair and skin disorders as signs of mitochondrial diseases. Paediatrics. 103:428-433 1999
58. A.E. Yücel, H. Kart-Köseoglu, B. Demirhan, et al.: Cholesterol crystal embolization mimicking vasculitis: success with corticosteroid and cyclophosphamide therapy in two cases. Rheumatol Int. 26:454-460 2006
59. B.D. Sharma, S.R. Kabra, B. Gupta: Symmetrical peripheral gangrene. Trop Doct. 34:2-4 2004
60. B. Planchon, E. Becker, P.H. Carpentier, et al.: Acrocyanosis: changing concepts and nosological limitations. J Mal Vasc. 26:5-15 2001
61. M.R. Grima: Nursing case study: bilateral cervical sympathectomy for acrocyanosis. Nurs Times. 71:1850-1852 1975
62. H. Levesque: Classification of erythermalgia [French]. J Mal Vasc. 21:80 1996
63. C. Mørk, K. Kvernebo: Erythromelalgia: a mysterious condition?. Arch Dermatol. 136:406 2000
64. C. Mørk, C.L. Asker, E.G. Salerud, et al.: Microvascular arteriovenous shunting is a probable pathogenetic mechanism in erythromelalgia. J Invest Dermatol. 114:643 2000
65. C. Mørk, O.M. Kalgaard, K. Kvernebo: Impaired neurogenic control of skin perfusion in erythromelalgia. J Invest Dermatol. 118:699 2002
66. C. Mørk, O.M. Kalgaard, B. Myrvang, et al.: Erythromelalgia in a patient with AIDS. J Eur Acad Dermatol Venereol. 14:498 2000
67. C. Mørk, K. Kvernebo, C.L. Asker, et al.: Reduced skin capillary density during attacks of erythromelalgia implies arteriovenous shunting as pathogenetic mechanism. J Invest Dermatol. 119:949 2002
68. R.C. Littleford, F. Khan, J.J. Belch: Skin perfusion in patients with erythromelalgia. Eur J Clin Invest. 29:588 1999
69. M.D. Davis, F. Wilkins, T.W. Rooke: Between episodes of erythromelalgia: a spectrum of colors. Arch Dermatol. 142:1085 2006
70. R.H. Cook-Norris, M.M. Tollefson, A.E. Cruz-Inigo, et al.: Pediatric erythromelalgia: a retrospective review of 32 cases evaluated at Mayo Clinic over a 37-year period. J Am Acad Dermatol. 66 (416)2012
71. L. McCarthy, L. Eichelberger, E. Skipworth, et al.: Erythromelalgia due to essential thrombocythemia. Transfusion. 42:1245 2002
72. L.M. Coppa, K.S. Nehal, J.W. Young, et al.: Erythromelalgia precipitated by acral erythema in the setting of thrombocytopenia. J Am Acad Dermatol. 48:973 2003
73. Erythromelalgia in a patient with idiopathic thrombocytopenic purpura (letter). Br J Dermatol. 1 48G.P. Thami, M. Bhalla: Erythromelalgia induced by possible calcium channel blockade by ciclosporin. BMJ. 326:910 2003
74. C.K. Dolan, M.A. Hall, G.W. Turlansky: Secondary erythermalgia in an HIV-1-positive patient. AIDS Read. 113:91 2003
75. K.B. Reed, M.D. Davis: Incidence of erythromelalgia: a population-based study in Olmsted County, Minnesota. J Eur Acad Dermatol Venereol. 23:13 2009
76. J. Schechner: Red skin re-read. J Invest Dermatol. 119:781 2002
77. M.D.P. Davis, P. Sandroni, T.W. Rooke, et al.: Erythromelalgia: vasculopathy, neuropathy, or both? A prospective study of vascular and neurophysiologic studies in erythromelalgia. Arch Dermatol. 139:1337 2003
78. N. Charkoudian: Skin blood flow in adult human thermoregulation: how it works, when it does not, and why. Mayo Clin Proc. 78:603 2003
79. B. Kazemi, S.M. Shooshtari, M.R. Nasab, et al.: Sympathetic skin response (SSR) in erythromelalgia. Electromyogr Clin Neurophysiol. 43:165 2003
80. M.D. Davis, T.W. Rooke, P. Sandroni: Mechanisms other than shunting are likely contributing to the pathophysiology of erythromelalgia. J Invest Dermatol. 115:1166 2000
81. K. Orstavik, C. Weidner, R. Schmidt, et al.: Pathological C-fibres in patients with a chronic painful condition. Brain. 126:567 2003
82. R.B. Layzer: Hot feet: erythromelalgia and related disorders. J Child Neurol. 16:199 2001
83. Y. Sugiyama, S. Hakusui, A. Takahashi, et al.: Primary erythromelalgia: the role of skin sympathetic nerve activity. Jpn J Med. 30:564 1991
84. J.P. Drenth, W.H. Finley, G.J. Breedveld, et al.: The primary erythermalgia-susceptibility gene is located on chromosome 32. Am J Hum Genet. 68:1277 2001
85. Y. Yang, Y. Wang, S. Li, et al.: Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 41:171 2004
86. T.R. Cummins, S.D. Dib-Hajj, S.G. Waxman: Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci. 24:8232 2004
87. J.J. Michiels, R.H. te Morsche, J.B. Jansen, et al.: Autosomal dominant erythermalgia associated with a novel mutation in the voltage-gated sodium channel alpha subunit Nav1.7. Arch Neurol. 62:1587 2005
88. C. Han, S.D. Dib-Hajj, Z. Lin, et al.: Early- and late-onset inherited erythromelalgia: genotype-phenotype correlation. Brain. 132:1711 2009
89. M. Estacion, S.G. Waxman, S.D. Dib-Hajj: Effects of ranolazine on wild-type and mutant hNav1.7 channels and on DRG neuron excitability. Mol Pain. 6:35 2010
90. S.G. Waxman, S. Dib-Hajj: Erythermalgia: molecular basis for an inherited pain syndrome. Trends Mol Med. 11:555 2005
91. J.P. Drenth, R.H. Te Morsche, S. Mansour, et al.: Primary erythermalgia as a sodium channelopathy: screening for SCN9A mutations: exclusion of a causal role of SCN10A and SCN11A. Arch Dermatol. 144:320 2008
92. P.J. van Genderen, I.S. Lucas, R. van Strik, et al.: Erythromelalgia in essential thrombocythemia is characterized by platelet activation and endothelial cell damage but not by thrombin generation. Thromb Haemost. 76:333 1996
93. R. Kurzrock, P.R. Cohen: Erythromelalgia: review of clinical characteristics and pathophysiology. Am J Med. 91:416 1991
94. M.D. Davis, T. Rooke: Erythromelalgia. Curr Treat Options Cardiovasc Med. 4:207 2002
95. M.D. Davis, P. Sandroni: Lidocaine patch for pain of erythromelalgia. Arch Dermatol. 138:17-19 2002
96. P. Sandroni, M.D. Davis: Combination gel of 1% amitriptyline and 0.5% ketamine to treat refractory erythromelalgia pain: a new treatment option?. Arch Dermatol. 142:283 2006
97. A. Almahameed, D.S. Pinto: Pernio (chilblains). Curr Treat Options Cardiovasc Med. 10:128-135 2008