Capítulo 22 - Tumores y Malformaciones Vasculares
22.1 Introducción
Las anomalías vasculares tienen un aspecto similar, tanto si se localizan en la piel, como cuando se desarrollan en las mucosas o en las vísceras. Suelen ser lesiones planas o prominentes y con varios tonos de azul, rosa o rojo. Durante siglos, los profanos y los propios médicos llamaron a las anomalías vasculares “marcas de nacimiento (nevus)”utilizando nombres familiares de comida (“fresa”, “cereza”) o de bebidas (“vino de Oporto”).
Con la introducción de la histopatología a mediados del siglo XIX, estas anomalías se conocieron como angiomas. Durante los 100 años siguientes, los términos descriptivos e histológicos eran cada vez más confusos y las distintas clasificaciones no lograban diferenciar entre diferentes tipos de lesiones vasculares.
En 1982 Mulliken y Glowacki clasificaron las proliferaciones vasculares en “hemangiomas“y “malformaciones vasculares“ en base a los datos anatomoclínicos y la historia natural de las mismas. En 1996 la International Society for the Study of Vascular Anomalies (ISSVA) modifica la clasificación y las divide en tumores y malformaciones vasculares.
22.2 Clasificación De LasAnomalías Vasculares
Tabla 1
(Ver Clasificación completa en el apéndice final )

22.3 Tumores Vasculares
22.3.1 Hemangiomas
Los hemangiomas son los tumores benignos más frecuentes de la infancia, con una incidencia entre 4-10% de los menores de un año y de 2,5% en los recién nacidos, siendo más frecuentes en los prematuros. Son más frecuentes en las niñas, con una proporción que varía entre 6:1 a 2:1, con respecto a los niños, excepto en los prematuros en los que la proporción es de 1:1.
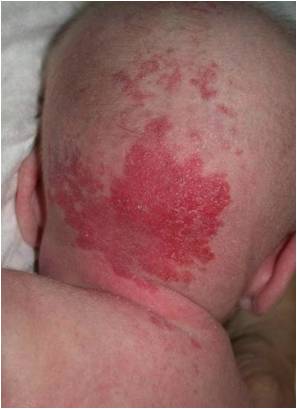
Los hemangiomas son una neoplasia vascular benigna, originada en las células endoteliales. Son los tumores de partes blandas más frecuentes en la infancia. Son lesiones caracterizadas por un período de crecimiento, seguido por uno de estabilización e inactividad, para finalmente involucionar y desaparecer espontáneamente en la mayoría de los casos. Hay una fase de crecimiento rápido que dura entre tres a diez meses, luego involucionan a partir de comienzos del segundo año, fase que puede durar entre dos a diez años. Aproximadamente 50-60% de los hemangiomas ha desaparecido a los 5 años de edad, 70% a los 7 años, 95-97% a los 10-12 años.
Esta característica los diferencia de las malformaciones vasculares, que son anomalías de los vasos sanguíneos y linfáticos, presentes desde el nacimiento, y persistentes por toda la vida y, por lo tanto, carentes de la evolución habitual de proliferación-reposo-involución, descrita para los hemangiomas.
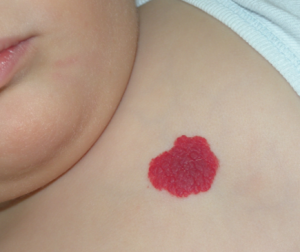
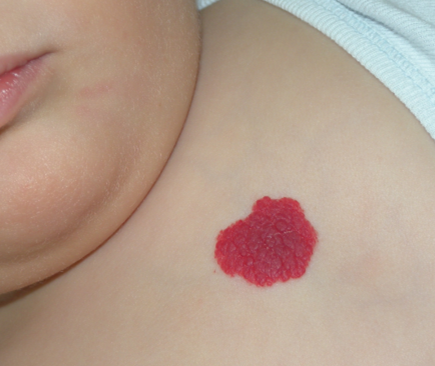
Fig. 1 Hemangioma
Los hemangiomas infantiles se pueden clasificar clínicamente según dos criterios: la profundidad de los vasos afectados y el patrón de forma-distribución.
En base a la profundidad hay tres tipos de hemangiomas cutáneos: superficiales, profundos y mixtos, siendo los primeros los más frecuentes.
1.- Hemangioma superficial (60%). Se ubica en la dermis superficial. Se presenta como unaelevación o pápula, o bien como una placa o nódulo rojo brillante, bien delimitado, firme y de superficie lisa o abollonada.
2.- Hemangioma profundo (15%). Se localiza en la dermis profunda y/o tejido subcutáneo. Corresponde a una masa o nódulo blando, mal delimitado, de color piel o tinte azulado.
3.- Hemangioma Mixto (22%). Combinación de ambas formas anteriores.
Los hemangiomas de cabeza, cuello y región lumbosacra pueden asociarse a lesiones estructurales subyacentes. (entre un 20-31% de los grandes hemangiomas faciales segmentarios lo presentan).
22.3.1.1 Localización
El sitio de presentación más frecuente es cabeza y cuello (60%), seguido por tronco (25%) y extremidades (15%). Además, pueden presentarse en casi cualquier órgano interno. Diversos estudios encontraron una incidencia de 11,5% de hemangiomas viscerales asociados a hemangiomas cutáneos. Los lugares de presentación más frecuentes fueron la laringe, hígado y el tracto gastrointestinal.La mayoría de los hemangiomas son únicos (80%), aunque entre un 15-30% de los niños pueden tener lesiones múltiples.
22.3.1.2 Evolución
En la evolución se reconocen tres fases:
1.-Fase de crecimiento: desde el nacimiento hasta el primer año de vida.
2.- Fase de estabilización: desde el primer año hasta los dos años de edad.
3.- Fase de involución: Desde los dos años hasta los 7 años
La lesión inicial puede ser una mácula pálida, eritematosa o telangiectásica, con o sin halo pálido, un grupo de pápulas rojas brillantes o una mácula-pápula azulada. Los hemangiomas superficiales evolucionan a nódulos con forma de cúpula, de color rojo vivo o placas de consistencia elástica que empalidece parcialmente con la compresión. Los hemangiomas profundos son de color piel o azulados, algo más compresibles y en su superficie pueden observarse telangiectasias,vasos y venas pequeñas. Estos últimos pueden cambiar de tamaño y color con el llanto o la actividad.
El primer signo de involución es la decoloración grisácea, la cual comienza desde el centro de la lesión, junto con la pérdida de consistencia y el aplanamiento de esta. La regresión de los profundos es más difícil de apreciar, pero la progresión es similar. La rapidez y el grado de involución depende de cada hemangioma, pero hay localizaciones más difíciles, tales como los labios, la parótida, o la punta nasal (nariz de Cyrano). Aproximadamente, un 40% de los niños presenta algún cambio cutáneo residual, siendo los más frecuentes atrofia, piel redundante, telangiectasias, decoloración y cicatrices. La involución espontánea del hemangioma, permite que el 50% de los casos, la piel recupere una apariencia normal, sin dejar cicatrices o manchas residuales.
22.3.1.3 Complicaciones
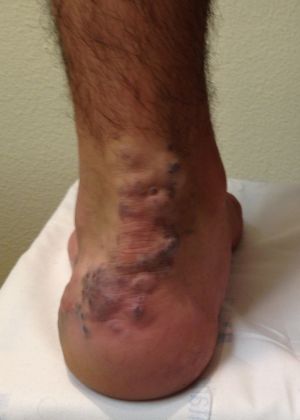
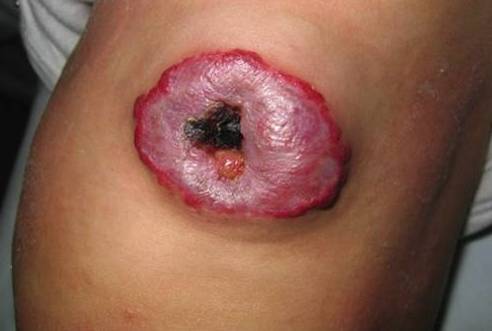
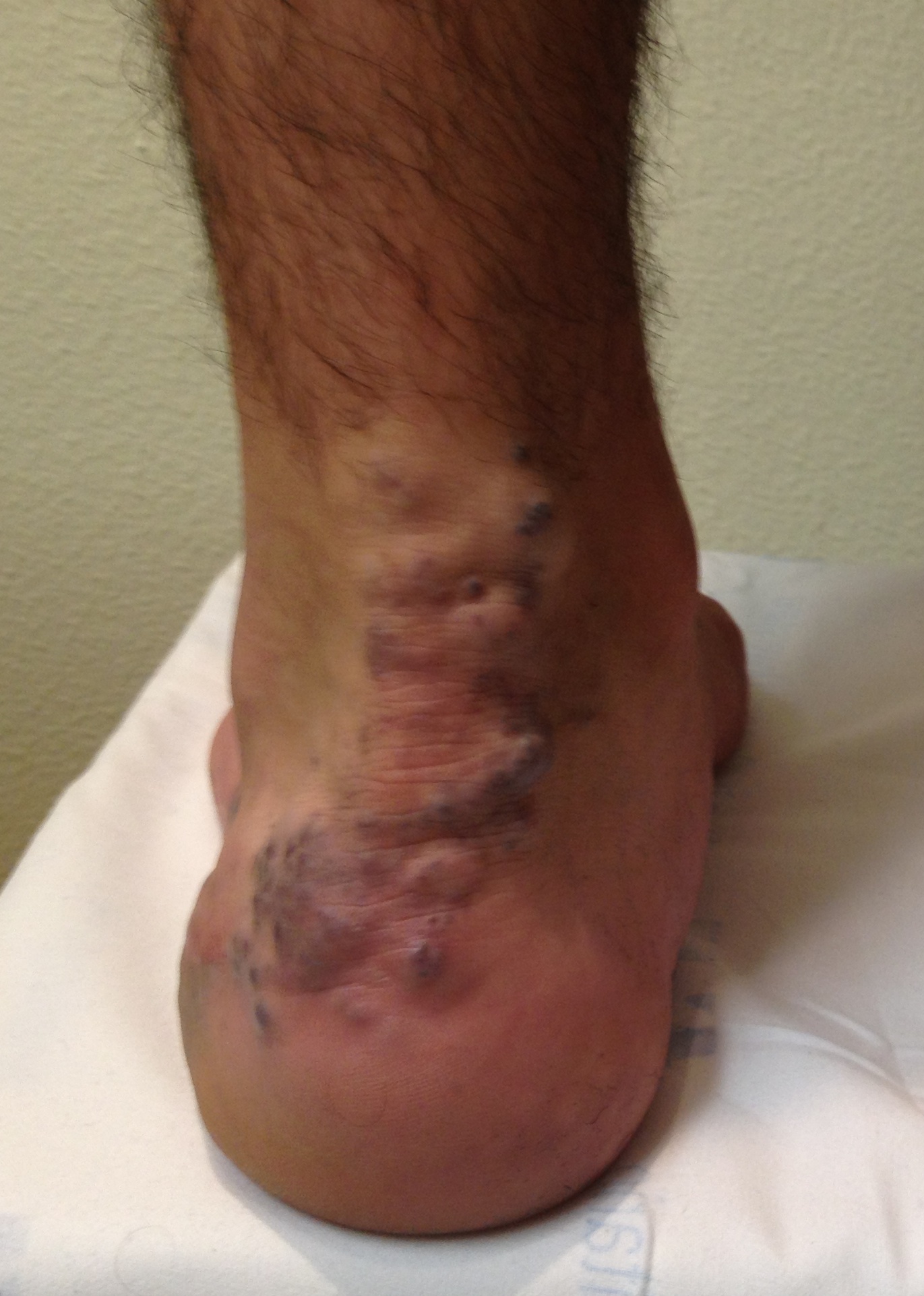
La ulceración es la complicación más común, presentándose en 5-13% de los casos, pudiendo ser muy dolorosa, especialmente en la región perioral y perianal. Estas lesiones pueden sangrar o sobreinfectarse. El sangrado cede con la compresión, aunque se han publicado casos de hemorragias masivas de difícil manejo. La sobreinfección generalmente estálimitada a la piel, pero puede complicarse con una celulitis u osteomielitis. Signos de alerta son: exudación de la piel circundante, supuración, calor, sensibilidad, dolor y fiebre. El agente más frecuentemente involucrado es el Estreptococo B-hemolítico grupo A. La ulceración puede dejar cicatrices que pueden ser desfigurantes, dependiendo del tamaño del hemangioma.
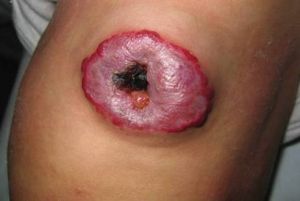
Fig. 2 Hemangioma Ulcerado
Los hemangiomas peri-orbitarios, particularmente los de párpado superior, pueden generar compromiso visual. El problema más común es el astigmatismo por la compresión del globo óptico o expansión del hemangioma en el espacioretrobulbar.Otras complicaciones son ptosis con daño corneal, ambliopía y estrabismo.
El compromiso respiratorio es consecuencia de una obstrucción de la vía aérea, debido a hemangiomas en las fosas nasales, orofaringe o región laringotraqueal. Otra complicación importante deriva del tamaño y rápido crecimiento de algunos hemangiomas, especialmente en cabeza y cuello, produciendo deformaciones y problemas psicológicos para los padres y la familia.
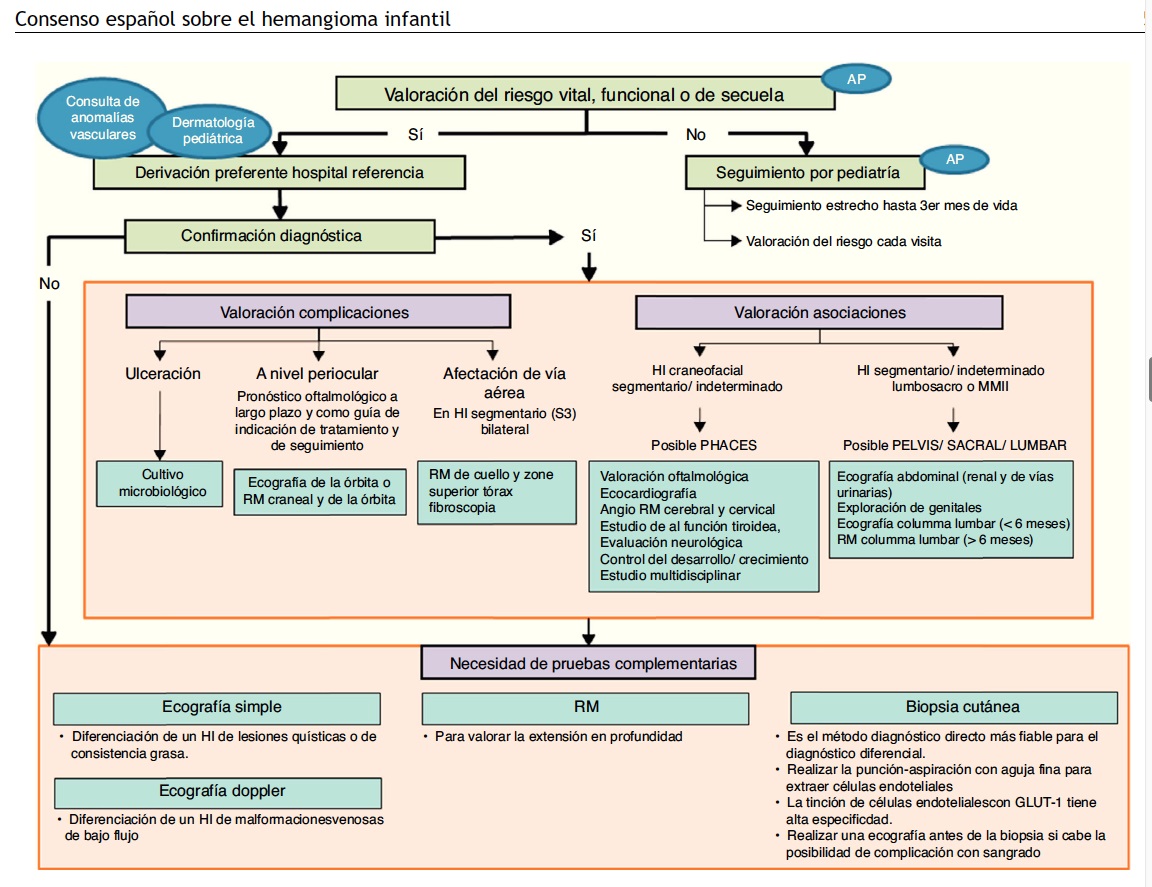
22.3.1.4 Estudios de Imagen
La ecografía con Doppler color es útil en casos en que se tiene duda con respecto al diagnóstico diferencial con malformaciones vasculares; sin embargo, la diferenciación es difícil y requiere de un especialista con experiencia en el diagnóstico de lesiones vasculares. También la ecografía debe ser solicitada para descartar hemangiomas hepáticos, cuando el niño presenta múltiples hemangiomas cutáneos (más de cinco).
Dado que los hemangiomas de la línea media pueden ser marcadores de disrafia espinal, se debe pedir una evaluación radiológica consistente en una ecografía, si el niño tiene menos de seis meses de edad, y en una resonancia nuclear magnética (RNM) si es mayor. También debe considerarse la realización de una RNM cuando se presentan grandes hemangiomas cervicofaciales y torácicos, ya que un 5-10% de los casos, especialmente en niñas, puede estar asociado a una anomalía estructural.
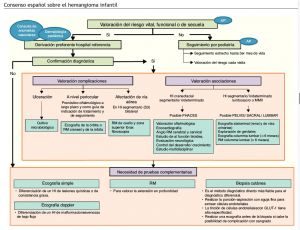
Fig. 3 Consenso español sobre hemangioma infantil
22.3.1.5 Diagnóstico DiferenciaL
Se debe establecer especialmente con las malformaciones vasculares. Estas, a diferencias de los hemangiomas, son errores en el desarrollo de los vasos capilares, venosos, linfáticos o arteriales, que siempre están presentes en el momento del nacimiento, crecen proporcionalmente con el niño y no involucionan.
Una excepción serian las “manchas salmón” de la frente, párpados o nuca, que desaparecen al año de vida y son consideradas como vasos sanguíneos fetales persistentes y o malformaciones capilares.
Los hemangiomas profundos deben diferenciarse de los lipomas, gliomas, sarcomas o quistes o herniaciones congénitas de estructuras neurales.
También debe realizarse un diagnóstico diferencial con gliomas nasales, quistes dermoides, miofibromatosisinfantil, neuroblastomas, neurofibromas plexiformes, pilomatricomas,lipomas y otros sarcomas, si bien las técnicasde imagen suelen ser suficientes para esclarecer eldiagnóstico. A su vez, los HI multifocales deben diferenciarsede la linfangioendoteliomatosis multifocal, elsíndrome de Bean y la histiocitosis de células de Langerhans.Por último, los HI superficiales pueden confundirse conhemangiomas en penachos, hemangioendoteliomas kaposiformes,hemangiopericitomas o angiosarcomas.
22.3.1.6 Tratamiento
Dada la evolución natural de los hemangiomas, la mayoría sólo requiere de seguimiento y observación por parte del médico, debiendo ser tratados quirúrgicamente aproximadamente solo el 10% de todos los casos. Los hemangiomas pueden causar mucha ansiedad por parte de los padres, por lo que es fundamental explicarles en quéconsiste esta afección y cuál es su comportamiento. Es importante fortalecer la relación médico-paciente, especialmente en aquellos de localización facial o de gran tamaño, de manera que los padres puedan enfrentar sus temores y la estigmatización social a la cual pueden estar sometidos.
La observación clínica es la conducta a seguir en la mayoría de los hemangiomas, debido a que la regresión espontánea es la regla. La tasa de regresión es variable y no esta relacionada con el tipo de hemangioma, ni sexo del paciente, ni tamaño, ni sitio afectado. En la fase de crecimiento, el control debe ser mensual para detectar complicaciones como la ulceración o hemorragia.
En las fases estacionarias o involutivas, los controles deben postergarse cada tres a seis meses. Los signos de involución recordemos que son cambio de color, de rojo brillante a pálido, aparición de áreas blanquecinas en el centro del hemangioma y consistencia más blanda.
Los siguienteshemangiomas requieren tratamiento especial:
*Hemangioma periorificial.
*Hemangioma parotídeo y o de vísceras comprometidas por el hemangioma.
*Hemangioma ulcerado.
*Hemangioma extenso que abarque una zona mayor al 5% corporal.
*Hemangioma que comprometa la vía aérea.
*Hemangioma que afecte en forma importante la estética.
*Hemangioma múltiple neonatal.
Los fundamentos para tratar un hemangioma deben ser el prevenir o revertir cualquier complicación, especialmente aquellas de riesgo vital; prevenir desfiguramientos residuales permanentes, minimizar los efectos psicológicos en el paciente, padres y familiares; prevenir el crecimiento excesivo y la ulceración que pueden dejar cicatrices.
Todos los hemangiomas pasan por las tres etapas de desarrollo descritas, sin embargo, existe gran heterogeneidad en la velocidad y duración de la etapa de crecimiento, sin que exista un marcador que nos permita distinguir cuál de ellos tendráun crecimiento acelerado o mayor riesgo de complicaciones. Aún así, existe consenso en que la conducta terapéutica es función de los siguientes factores:
1.-Localización. Sin duda que la ubicación en la vía aérea es una emergencia médica que requiere de tratamiento enérgico, inmediatamente efectuado el diagnóstico. De igual manera la localización peri-ocular necesita tratamiento oportuno, asícomo todas las ubicaciones peri orificiales: hemangiomas de la región genital, anal, ótica o nasal. Los hemangiomas ubicados en cualquiera de los pliegues (cervical, retroauricular, axilar, inguinal, poplíteo) tienen un mayor riesgo de ulceración, como también los ubicados en la zona del pañal. La localización facial y en otras zonas de alto valor estético (hombros, cuello, zona del escote y dorso alto en las niñas), pueden tener mejor resultado con tratamiento que con la evolución espontánea.
2.-Velocidad de crecimiento. Como ya se mencionó, la velocidad de crecimiento es muy variable entre un hemangioma y otro; por lo tanto, el pediatra debe evaluar en el control periódico del niño el incremento de tamaño o volumen que presenta la lesión, si dobla su medida en el transcurso de un mes, requiere de tratamiento.
3.-Presencia de complicaciones.Hemangiomas ulcerados, con o sin sobreinfección secundaria o sangrado de repetición son casos que exigen un tratamiento urgente. También requieren tratamiento adecuado -y nunca demorado- la hemangiomatosis diseminada, especialmente con compromiso visceral. Algunos hemangiomas, especialmente los de gran tamaño, pueden atrapar plaquetas y consumir factores de coagulación, con anemia moderada o severa del paciente.
La respuesta al tratamiento en estos casos es mejor mientras más precoz se inicie este; es decir, cuando los factores angiogénicos son claramente predominantes en plena etapa proliferativa. No debe postergarse el tratamiento en espera de una resolución espontánea, ya que ésta sólo ocurrirálenta y tardíamente en la etapa involutiva.
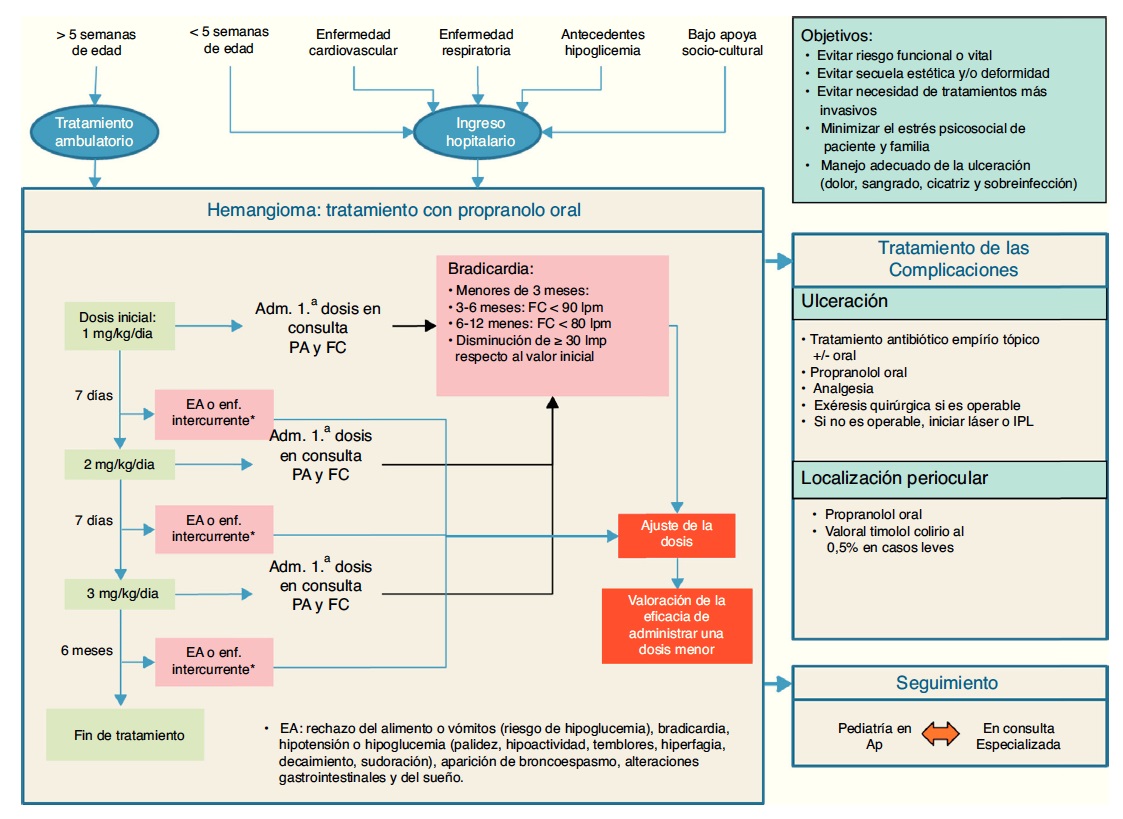
Hay distintas modalidades terapéuticas, las cuales deben aplicarse según la edad del menor, el tamaño, ubicación y etapa de crecimiento o involución del hemangioma, para lo cual debe ser derivado al especialista con experiencia en manejo de lesiones vasculares en niños.
El tratamiento médico de elección fueron los corticoides oraleshasta el año 2008, a pesar de sus efectos secundarios a dosis altas y a la ausencia de respuesta al tratamiento en un tercio de los casos.
En la actualidad, el propranolol por vía oral se considera el tratamiento de elección (1 mg/kg/día durante la primera semana, 2 mg/kg/día durante la segunda semana y 3 mg/kg/día durante 6 meses) ya que es el único aprobado para dicha indicación.La eficacia del propranolol es superior a la de cualquier otro tratamiento, desde el inicio y en cualquier localización corporal. Los efectos secundarios de los betabloqueantes son bien conocidos: 1) cardiovasculares (bradicardia e hipotensión); 2) bronquiales (reducción del tono broncodilatador e incremento de la resistencia de las vías aéreas de mediano calibre); 3) metabólicos (hipoglucemia); 4) renales (reducción del filtrado glomerular), y 5) sistema nervioso central (posible afectación de la memoria, la calidad del sueño, el estado de ánimo y las funciones psicomotoras).
Hemangiomas grandes de difícil manejo, y refractarios a otras terapias, han sido tratados con interferón alfa, con buenos resultados. Sin embargo, debe ser estrictamente monitorizado, ya que se han observado efectos adversos neurológicos, tales como letargia, lentitud psicomotora, confusión y paresia espástica.
El timolol, en forma tópica, se ha mostrado efectivo en lesiones superficiales.
Se ha investigado el uso del láser en el manejo de los hemangiomas, sin embargo, por la penetración que tienen en la piel, solamente están indicados en los hemangiomas ulcerados, hemangiomas superficiales muy pequeños o para tratar telangiectasias que aparecen en la evolución.
22.3.1.6.1 Indicación de Tratamiento Quirúrgico
La cirugía de los hemangiomas debe reservarse solo para aquellos casos que han fallado al tratamiento y tienen riesgo vital. Obstrucción visual y o glótica que no responde a tratamiento sistémico. Ulceración o hemorragia que no responde a medidas locales o sistémicas.
La principal utilidad de la cirugía escuando ya ha concluido la fase de involución, ante la presencia de lesiones residuales y cicatrices, para la reparación de secuelas desfigurantes, cicatrices y residuos lipomatosos que dejan los hemangiomas de gran tamaño. En general, la cirugía reparadora se programa en la edad preescolar, para que el ingreso del niño al colegio sea sin problemas psicológicos derivados de las secuelas del hemangioma.
22.3.1.6.1.1 Tratamiento del Hemangioma Ulcerado
Hay varios aspectos involucrados en el manejo de la ulceración, ya que se debe tratar la herida, la sobreinfección y el dolor. Además del tratamiento específico del hemangioma, para la herida se recomienda el uso de barrerasprotectora como óxido de zinc, parches, antibióticos tópicos y apósitos. Si hay signos de sobreinfección, debe tratar de objetivarse mediante un cultivo para identificar el germen involucrado y su sensibilidad para dar el antibiótico adecuado, el cual se puedes usar por vía oral y local.
El dolor debe ser considerado como un síntoma adverso y que causa deterioro clínico del paciente, éste provoca irritabilidad, anorexia, insomnio en los niños y gran ansiedad por parte de los padres. El manejo del dolordebe ser agresivo, en especial al manipular laslesiones, como con el cambio de apósitos y durantela limpieza de las mismas. Se puede usar paracetamol como primer analgésico, el cual puede combinarse con codeína en lesiones muy dolorosas.
Fig. 4 Algoritmo del tratamiento del hemangioma infantil
22.3.1.7 Conclusión
Los hemangiomas son tumores vasculares que deben diferenciarse de las malformaciones. Estos tienen un curso habitual que tiende a la involución y regresión completa. Sin embargo, alrededor de un 10% de los casos va a requerir tratamiento, ya sea por su localización, crecimiento acelerado o presencia de complicaciones. El tratamiento debe ser lo más precoz posible para un mejor resultado, y debe ser elegido para cada paciente en particular. El objetivo final es que la regresión del hemangioma sea completa y sin secuelas físicas ni psicológicas.
22.3.2 Otros Tumores Vasculares
22.3.2.1 Angioma Encopetado
Descrito por primera vez a finales de los años 80. Es un tumor vascular benigno caracterizado por copetes de capilares en dermis. Se presentan en tronco y extremidades y puede asociarse al Fenómeno de Kasabach_Merritt (trombocitopenia, hipofrinogenemia elevación de productos de la degradación de la fibrina y del Dímero D). Su aspecto puede variar desde nódulos anulares eritematosos a placas con o sin hipertricosis.
22.3.2.2 Hemangioendotelioma Kaposiforme
Muestra predilección por tórax, hombro y desde ingle hasta pierna.. Se distribuye por igual entre ambos sexos y se asocial al Fenómeno de Kasabach-Merritt (trombocitopenia, hipofrinogenemia elevación de productos de la degradación de la fibrina y del Dímero D). Cuando involucionan muestran un comportamiento histológico y clínico muy diferente de los hemangiomas. El tratamiento depende de la localización y las características radiológicas y puede incluir esteroides, quimioterapia, interferón , antifibrinolíticos, antiagregantes y embolización.
22.3.2.3 Granuloma Piógeno (Hemangioma Capilar Lobular)
Son lesiones adquiridas, papulares, pequeñas y que pueden sangrar en piel, o con menor frecuencia en mucosas, y que aparecen en la infancia. Tienen preferencia por la región cervicofacial pero pueden encontrarse también tórax y extremidades. Pueden ser tratadas mediante extirpación, cauterización, crioterapia, láser de CO2 o escleroterapia.
22.3.2.4 Sarcoma De Kaposi
Se observa con frecuencia asociada al SIDA pero no de forma exclusiva. Es una neoplasia vascular infrecuente que comienza como máculas azuladas violáceas que se transformar paulatinamente en placas, pápulas y nódulos. Es multifocal, es decir, se presenta simultáneamente en varias zonas all mismo tiempo sin que se traten de metástasis.Parece tener una etiológia vírica. Se trata mediante fármacos antivíricos, antiangiogénicos e inmunosupresores. En el caso de estar asociado al SIDA los antirretrovirales detienen su crecimiento
22.3.3 Tumores Vasculares Primarios
Los tumores de la pared de arterias y venas son raros, apenas se han registrado poco mas de un centenar de casos.
La arteria donde mas casos se han descrito es la aorta. En las venas el tipo mas frecuente es el leiomiosarcoma y afecta sobre todo a la vena cava inferior (60%)
Se clasifican (Whright 1985) en : intimales (70%) y murales. El diagnóstico mas preciso es inmunohistoquímico.
El diagnóstico es difícil y tardío y hasta en un 60% de los casos existen metástasis cuando se diagnostican y menos del 5% se diagnostican preoperatoriamente.
La clínica es inespecífica y la mayoría tan solo presentan un síndrome constitucional: adelgazamiento, cansancio, nauseas,… aunque a veces pueden debutar con una embolia visceral o en extremidades ( especialmente los tumores intimales). La mayoría de los tumores vasculares venosos se presentan en mujeres (>80%) y edad media. La clínica de estos tumores está en relación con las metástasis y las secuelas de la obstrucción de la cava (edema en miembros inferiores)
En general, los estudios radiológicas sirven para detectar un defecto o irregularidad en la pared del vaso pero no permite diferenciar una placa de ateroma de un tumor vascular. Posiblemente la RNM es la técnica que mas nos pueda acercar a un diagnóstico preoperatorio.
El tratamiento de elección es la resección quirúrgica y la sustitución por una prótesis. El pronóstico es malo y la supervivencia media es de 14 meses. La respuesta a la quimioterapia y radioterapia es también mala.
22.4 Malformaciones Vasculares
Las malformaciones vasculares pueden ser errores localizados o difusos producidos durante el desarrollo embrionario. La clasificación se basa en el tipo de vaso predominante: capilares, linfáticos, venas, arterias o cualquier combinación de ellos. El término vasculogeniase refiere al proceso por el cual los precursores endoteliales de origen mesodérmico se alinean para formar vasos primitivos.
Se sabe que las diferencias entre arterias y venas se establecen al principio del desarrollo embrionario. Las células endoteliales arteriales expresan el ligando transmenbrana efrina B-12, mientras que las venas expresan el receptor Eph-B4. El término angiogenia se refiere a la formación de nuevos vasos a partir de vasos ya existentes. Los distintos tipos de malformaciones vasculares se pueden contemplar como un desarrollo incorrecto en alguna etapa de la vasculogenia o de la angiogenia.
La mayoría de las malformaciones vasculares son esporádicas, pero algunas se heredan con un patrón autonómico dominante. Se han identificado los genes causales de varios trastornos familiares, abriendo la puerta al conocimiento de los mecanismos básicos implicados en su patogenia. Los estudios moleculares indican que las anomalías vasculares están causadas por disfunciones del proceso de señalización que regulan la proliferación, apoptosis, diferenciación, maduración y adhesión de las células vasculares.
Por otra parte las malformaciones vasculares se consideran anomalías congénitas de crecimiento lento asociados a cortocircuitos arteriovenosos. Las malformaciones vasculares se subdividen (Modified Hamburg Classification) en: a) una clasificación primaria basada en el defecto vascular predominante (capilares, linfáticos, venas, arterias o cualquier combinación de ellos) y b) una subclasificación embriológica basada en la anatomía y estadio evolutivo.
22.4.1 Malformaciones Capilares
22.4.1.1 Patogenia
Están integradas por vasos de tamaño que varia desde capilar hasta venular y se localizan en la dermis superficial. Los estudios inmunohistoquímicos han demostrado una disminución de las fibras nerviosas circundantes normales.
La malformaciones capilares constituyen la mayoría de las marcas vasculares de nacimiento, denominadas antes naevus flammeus neonatorum. Estas manchas de color rosa pálido aparecen en el 50% de los recién nacidos y se conocenpopularmente como “beso de ángel”(cuando se localiza en la frente, párpados y labio superior) y “picotazo de cigüeña“(en la región de la nuca).
22.4.1.2 Clínica
En el siglo XIX se las conocía como “manchas en vino de Oporto”. Pueden ser localizadas o generalizadas y raramente son múltiples. Pueden presentarse en cualquier parte del cuerpo, siendo más llamativas las faciales.En los adultos, estas manchas adquieren un tono más oscuro y tienen tendencia al sobrecrecimiento fibrovascular nodular y de los tejidos blandos y del hueso subyacente. En la cara, se suele producir un aumento del labio y de la encía afectada y generalmente del maxilar y la mandíbula.
Las malformaciones capilares pueden ser indicativas de una anomalía estructural subyacente. El síndrome de Sturge-Weber incluye una malformación capilar facial con anomalías vasculares oculares y leptomeningeas ipsilaterales. Estas últimas pueden ocasionar convulsiones, hemiplejia contralateral y retraso psíquico y motor. El estudio oftalmológico es imprescindible para evitar complicaciones como el desprendimiento de retina, el glaucoma y la ceguera.
22.4.1.3 Tratamiento
Se tratan mediante láser pulsado sintonizable. En general, se produce una decoloración significativa en el 70% de los casos. La hipertrofia del hueso y de los tejidos blandos requieren de intervenciones quirúrgicas.
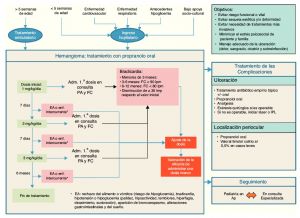
Fig. 5 Malformación Capilar
22.4.2 Malformaciones Linfáticas
22.4.2.1 Patogenia
Durante el siglo XX existióuna controversia sobre si los vasos linfáticos se originan a partir de venas preexistentes (teoría centrífuga) o si surgen independientemente y luego se conectan con las venas (teoría centrípeta). Las investigaciones más recientes apoyan el modelo de Sabin del desarrollo vascular centrífugo. Histológicamente, las malformaciones linfáticas están integradas por espacios vasculares repletos de un líquido eosinofílico rico en proteínas. Las paredes son de grosor variable y están formadas por elementos anómalos de músculo liso, junto con agregados linfocitarios.
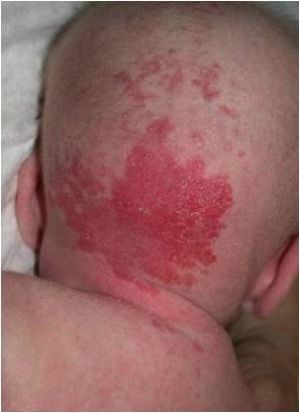
Fig. 6 Malformación Linfática
22.4.2.2 Clínica
Las malformaciones linfáticas suelen aparecer en el nacimiento o antes de los dos años de vida y se localizan principalmente en extremidades, axilas o en el tórax. Se han clasificado en microquísticas (“linfangioma”) o macroquísticas (“higroma cístico”).Con frecuencia la piel que las recubre es normal o con un tono azulado. La afectación dérmica se manifiesta como hoyuelos o fosetas dérmicas profundas.
Las malformaciones linfáticas de la frente y de la zona orbitaria causan proptosis, estrabismo, ambliopía y hemorragia intralesional recurrente. Las malformaciones linfáticas faciales son el origen más frecuente de macroquelia, macroglosia, macrotia y macromala (sobrecrecimiento de la mejilla o del hueso malar). Las malformaciones linfáticas cervicofaciales se asocian al sobrecrecimiento del cuerpo mandibular lo que provoca una mordida abierta o cerrada. Por último, las malformaciones linfáticas que afectan a la vías aéreas superiores supraglóticas suelen precisar una traqueostomia inmediata.
22.4.2.3 Tratamiento
Las dos complicaciones principales de las malformaciones linfáticas son la hemorragia y la infección intralesional. La hemorragia se aprecia por un cambio en la coloración, acompañado de dolor. El tratamiento es sintomático. En casos muy extensos se puede valorar el tratamiento antibiótico, como prevención de una celulitis, complicación rara pero peligrosa.
Las dos estrategias de tratamiento se basan en la escleroterapia y la resección. Los productos esclerosantes más utilizados son etanol, sulfato de sodio de tetradecil y doxiciclina.
22.4.3 Malformaciones Venosas
22.4.3.1 Patogenia
Histológicamente están formadas por vasos anómalos espongiformes y de paredes delgadas. La inmunotinción del músculo liso con actina revela la presencia de agregados de leiomiocitos en las paredes vasculares. Presumiblemente esta anomalía es la que explica la tendencia a la expansión de los vasos anormales. El estudio histológico suele describir la formación de coágulos, varias fases de crecimiento fibrovascular intimal y la presencia de flebolitos patognomónicos.

Fig. 7 Malformación Venosa
22.4.3.2 Clínica
Las malformaciones venosas son lasmalformaciones vasculares más frecuentes. Se presentan al nacimiento, aunque no siempre sean clínicamente evidentes y no involucionan. Son de flujo lento y se manifiestan de muchas formas. Típicamente de color azulado, blandas y compresibles. Pueden ser localizadas o generalizadas dentro de una región anatómica. Aunque la mayoría de las malformaciones venosas se producen en la piel y los tejidos subcutáneos, también pueden afectar al músculo subyacente, vísceras abdominales y sistema nervioso central. No se debe emplear el término de hemangioma cavernoso para definir estas malformaciones venosas más extensas. Clínicamente se ven como tumoraciones o nódulos de color azul y fácilmente compresibles. Pueden aumentar de tamaño con el declive o con las maniobras de Valsalva.
Las malformaciones venosas presentan diferencias, según la región anatómica afectada:
Cabeza y cuello, suelen ser unilaterales y producir una asimetría facial.
Extremidades, pueden afectar a la piel o extenderse al músculo subyacente.
Intestino, las malformaciones venosas del sistema digestivo suelen cursar con hemorragias crónicas y anemia. Pueden afectar cualquier zona del aparato digestivo, siendo más frecuentes en el colon. A veces se asocian a malformaciones cutáneas, dando origen al denominado “Síndrome del nevus azul”.
Síndrome del nevus azul en tetina de goma. Es un trastorno poco frecuente que asocia malformaciones venosas cutáneas y digestivas. Puede presentar una herencia autonómica dominante. Las lesiones cutáneas son blandas, de color azul y crecen con la edad. Las lesiones digestivas asientan principalmente en intestino delgado.
22.4.3.3 Tratamiento
La escleroterapia como tratamiento de una malformación venosa estáindicado para paliar su aspecto, dolor o problemas funcionales. El ácido acetil salicílico a dosis bajas minimiza las flebotrombosis dolorosas. La resección suele ser eficaz en lesiones pequeñas y localizadas. En las malformaciones más diseminadas, la extirpación total es más difícil y suele precisar de escleroterapia previa. Por último, en las hemorragias digestivas puede requerirse control endoscópico o quirúrgico.
22.4.4 Malformaciones Arteriovenosas
22.4.4.1 Patogenia
La telangiectasia hemorrágica hereditaria (enfermedad de Rendu-Osler-Weber) es la primera anomalía vascular diagnosticada a nivel molecular. El trastorno comienza en los lechos capilares a medida que diminutas derivaciones capilares-venosas aparecen en la piel y membranas mucosas, pulmones, hígado y encéfalo, para tener su máxima expresividad entre los 20 y 40 años de edad. Los pacientes con telangiectasia hemorrágica hereditaria presentan telangiectasias mucocutáneas,derivaciones arteriovenosas cerebrales, malformaciones arteriovenosas pulmonares y anomalías vasculares hepáticas. Se han identificado dos anomalías en el cromosoma 9q que afectan ambas a la unión y señalización del factor de crecimiento transformante β, TGFβ. La THH-1 esta causada por una mutación de la endoglina, un gen que codifica una glucoproteina endotelial y la THH-2 esta causada por una mutación en la activina, una cinasas de tipo receptor.
22.4.4.2 Clínica
Las malformaciones arteriovenosas se localizan con mayor frecuencia en cabeza y cuello. Las malformaciones arteriovenosas cutáneas se pueden diagnosticar al nacer, aunque presentan un aspecto inofensivo. Durante la infancia se pueden confundir con un hemangioma, aunque con el tiempo, se hace evidente la falta de involución. Con frecuencia, los cambios hormonales durante la pubertad o mínimos traumatismos desencadenan la extensión. El color de la mancha se oscurece o aparece un tumor debajo. A la exploración física se aprecia un aumento de la temperatura, un frémito palpable y la auscultación de un soplo. Las manifestaciones cutáneas tardías incluyen alteraciones isquémicas, úlceras, dolor incoercible y hemorragias intermitentes.
22.4.4.3 Tratamiento
En las malformaciones arteriovenosas puede ser necesaria una embolización rápida cuando se produce el caso poco frecuente de una insuficiencia cardíaca de alto gasto secundaria a una malformación arteriovenosa. En el resto de los casos, se debe proceder a una observación de la malformación, y tratar aquellos casos en los que aparezcan complicaciones, procediendo a una embolización selectiva asociada o no a una extirpación quirúrgica posterior.
22.4.5 Malformaciones Vasculares Combinadas
Las malformaciones vasculares combinadas o complejas se asocian a un sobrecrecimiento de los tejidos blandos y óseos subyacentes. Muchos de estos trastornos se denominan con el nombre del médico que realizóuna descripción original. Se pueden clasificar en anomalías combinadas de flujo lento o de flujo rápido.
22.4.5.1 Anomalías de flujo lento
Síndrome de Klippel-Trenaunay es el epónimo más utilizado para la malformación capilar-linfática-venosa que se asocia a hipertrofia de tejidos blandos y hueso, generalmente de una o más extremidades y en ocasiones del tronco. En los recién nacidos, el componente es macular, pero con el tiempo las zonas manchadas aparecen sembradas con vesículas hemolinfáticas. Las venas se hacen cada vez más prominentes por insuficiencia valvular y en más del 50% de los pacientes existe hipoplasia linfática y produce linfedema. También es evidente la hipertrofia de la extremidad. El tratamiento: depende de la gravedad del cuadro clínico, pudiendo requerir una corrección de la longitud de la extremidad; mientras que el tratamiento de las malformaciones de las venas superficiales suele hacerse mediante esclerosis o cirugía.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fig. 8 Síndrome de Klippel-Trenaunay
22.4.5.2 Anomalías de flujo rápido
Síndrome de Parker Weber se describe como un rubor capilar con fístulas microarteriovenosas intramusculares y subcutáneas. Afectan sobre todo a las extremidades inferiores y se manifiestan desde el nacimiento. El miembro afectado suele presentar una mancha macular de color rosa y esta aumentado de tamaño. La presencia de un soplo a la exploración física confirma el diagnóstico. El tratamiento depende de los síntomas. Una embolización supraselectiva estáindicada en casos de síntomas isquémicos o dolorosos.
22.5 Bibliografía
1. Mulliken JB, Glowacki J: Hemangiomas and vascular malformations in infants and children: A classification based on endothelial characteristics. Plast Reconstr Surg 1982; 69:412.
2. Enjolras O, Mulliken JB: Vascular tumors and vascular malformations. Adv Dermatol 1998; 13:375.
3. Takahashi K, Mulliken JB, Kozakewich HPW, et al: Cellular markers that distinguish the phases of hemangioma during infancy and childhood. J Clin Invest 1994; 93:2357.
4. Gonzalez-Crussi F, Areyes-Mugica M: Cellular hemangiomas (“hemangioendotheliomas”) in infants: Light microscopic, immunohistochemical, and ultrastructural observations. Am J Surg Pathol 1991; 15:769.
5. Boon LM, Enjolras O, Mulliken JB: Congenital hemangioma: Evidence of accelerated involution. J Pediatr 1996; 128:329.
6. Finn MC, Glowacki J, Mulliken JB: Congenital vascular lesions: Clinical application of a new classification. J Pediatr Surg 1983; 18:894.
7. Dubois J, Patriquin HB, Garel L, et al: Soft tissue hemangiomas in infants and children: Diagnosis using Doppler sonography. AJR, Am J Roentgenol 1998; 171:247.
8. Pasquale-Castroviejo I, Viano J, Moreno F, et al: Hemangiomas of the head, neck and chest with associated vascular brain anomalies: A complex neurocutaneous syndrome. AJNR, Am J Neuroradiol 1996; 17:461.
9. Wang HU, Chen Z-F, Anderson DJ: Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 1998; 983:741.
10. Burrows PE, Laor T, Paltiel H, Robertson RL: Diagnostic imaging in the evaluation of vascular birthmarks. Dermatol Clin 1998; 16:455.
11. Berenguer B, Burrows PE, Zurakowski D, Mulliken JB: Sclerotherapy of craniofacial venous malformations: Complications and results. Plast Reconstr Surg 1999; 104:1.
12. Kohout MP, Hansen M, Pribaz JJ, Mulliken JBrteriovenous malformations of the head and neck: Natural history and management. Plast Reconstr Surg 1998; 102:643.
13. Samuel M, Spitz L: Klippel-Trenaunay syndrome: Clinical features, complications and management in children. Br J Surg 1995; 82:757.
14. Samuel M, Spitz L: Klippel-Trenaunay syndrome: Clinical features, complications and management in children. Br J Surg 1995; 82:757.
15. Medicina Vascular. Complemento de Braunwald. Tratado de Cardiología. 2ª Edición. Mark A. Greager, Joshua A. Beckman. Edt Elservier Saunders 2013.
16. Rutherford´s Vascular Surgery. 8Th Edition. Rutherford. Jack L. Cronenwett MD, K. Wayne Jhonston. Elservier Saunders 2014.
17. Baselga Torres et al. Consenso español sobre el hemangioma infantil. An Pediar (Barc). 2015. http://dx-doi.org/10.1016/j.anpedi.2015.10.004
| Tumores Vasculares | Malformaciones Vasculares | |||
|
Benignos Localmente agresivos o límites Malignos |
Simple | Combinadasº | De vasos mayores | Asociadas con otras anomalías |
|
Malformaciones Capilares Malformaciones Linfáticas Malformaciones Venosas Malformaciones Arteriovenosas* Fístulas Arteriovenosas * |
CVM, CLM LVM, CLVM CAVM* CLAVM* Otras |
Ver detalles | Ver lista | |
ºdefinido como dos o más malformaciones encontradas en una lesión
*lesiones de alto flujo
| TUMORES VASCULARES BENIGNOS |
| Hemangioma Infantil ( ver detalles más adelante) |
| Hemangioma Congénito |
| Rápidamente involutivo ( RICH) * |
|
No-involutivo ( NICH) |
| Parcialmente involutivo (PICH) |
| Angioma Encopetado *º |
| Hemangioma fusiforme |
| Hemangioma Epiteloide |
| Granuloma Piógeno ( Hemangioma Capitar Lobular ) |
| Otros |
| TUMORES VASCULARES LOCALMENTE AGRESIVOS O LÍMITES |
| Hemangioendotelioma Kaposiforme *º |
| Hemangioendotelioma Retiforme |
| Angioendotelioma Papilar intralimfático (PILA), Tumor de Dabska |
| Hemangioendotelioma Combinado |
| Sarcoma de kaposi |
| Otros |
| TUMORES VASCULARES MALIGNOS |
| Angiosarcoma |
| Hemangioendotelioma Epitelioide |
| Otros |
** algunas lesiones pueden estar asociadas a trombocitopenia y/o coagulopatía de consumo (ver detalles)
° algunos expertos creen que forman parte de un conjunto de entidades diferentes. Las lesiones vasculares proliferativas reactivas aparecen en tumores benignos
| MALFORMACIONES VASCULARES SIMPLES I |
| Malformaciones Capilares (CM) |
| Cuáneas y/o mucosas CM G |
| CM con hipertrofia de hueso y pates blandas |
| CM con anomalías SNC y/o oculares (síndrome Sturge-Weber) |
| CM of CM-AVM G |
| CM of MICCAP (microcefalia- malformación capilar) |
| CM of MCAP (megalencefalia - malformación capilar - polimicrogiria) |
| Telangiectasia |
| Telangiectasia Hereditaria hemorrágica (HHT) (diferentes tipos) G |
| Otras |
| Cutis marmorata telangiectasia congénita (CMTC) |
| Nevus simple / Parche de Salmon / “beso del angel”, “pico de cigüeña” |
| Otro |
Ir a G para ver genética
| MALFORMACIONES VASCULARES SIMPLES II |
| Malformaciones Linfáticas(LM) |
| Común (cística ) LM G |
| Macrocística LM |
| Microcística LM |
| Cística Mixta LM |
| Anomalía Linfática Generalizada (GLA) |
| LM en la enfermedad Gorham-Stout |
| LM tipo Canal |
| Linfedema Primario (diferentes tipos) G |
| Otras |
Algunas lesiones pueden estar asociadas a trombocitopenia y/o coagulopatia de consumo. Ir a G para ver genética
| MALFORMACIONES VASCULARES SIMPLES IIb |
| Linfedema Primario |
| Síndrome Nonne-Milroy G |
| Linfedema Primario Herediatario G |
| Linfedema-distiquiasis G |
| Hipotircosis-lifendema-telangiectasia G |
| Linfedema Primario con mielodisplasia G |
| Anomalia linfática primaria generalizada ( Síndrome linfangiectasia-linfedema de Hennekam ) G |
| Síndrome de Microcefalia con/o sin corioretirnopatía , linfedema o retraso mental G |
| Linfedema - atresia coanal G |
Ir a G para ver genética
| MALFORMACIONES VASCULARES SIMPLES III |
| Malformaciones Venosas (VM) |
| Cómun VM G |
| Familiar VM cutáneo-mucosa (VMCM) G |
| VM Síndrome del nevo gomoso azul G |
| Hipotircosis-lifendema-telangiectasia G |
| Malformación Cerebral cavernosa (CCM) (diferentes tipos)G |
| Otras |
Algunas lesiones pueden estar asociadas a trombocitonia y/o coagulopatía de consumo. Ir a G para ver genética
| MALFORMACIONES VASCULARES SIMPLES IV |
| Malformaciones Arteriovenosas (AVM) |
| Esporádica |
| En HHT G |
| En CM-AVMG |
| Otras |
| Fístula Arteriovenosa (AVF) ( Congénita ) |
| Esporádica |
| En HHT G |
| En CM-AVMG |
| Otras |
Ir a G para ver genética
| MALFORMACIONES VASCULARES COMBINADAS * | ||
| CM + VM | Capilar - malformación venosa | CVM |
| CM + LM | Capilar - malformación linfática | CLM |
| CM +AVM | Capilar - malformación arteriovenosa | CAVM |
| LM +VM | malformación linfática-venosa | LVM |
| CM + LM +VM | malformación capilar-linfática-venosa | CLVM |
| CM + LM +AVM | malformación capilar-linfática-arteriovenosa | CLAVM |
| CM + VM + AVM | malformación capilar-venosa-arteriovenosa | CVAVM |
| CM + LM + VM + AVM | malformación capilar-linfática-venosa-arteriovenosa | CLVAVM |
Definida como dos o más malformaciones vasculares encontradas en una lesión
| ANOMALIAS DE GRANDES VASOS | |
| Afectan a | Linfáticos |
| Venas | |
| Arterias | |
| Anomalías del | Origen |
| Trayecto | |
| Número | |
| Longitud | |
| Diámetro ( aplasia, hipoplasia, estenosis, ectasia/aneurisma) | |
|
Válvulas |
|
| Comunicación ( AVF ) | |
| Persistencia ( de vasos embrionarios ) | |
| MALFORMACIONES VASCULARES ASOCIADAS A OTRAS ANOMALÍAS | |
|
Síndrome Klippel-Trenaunay: CM + VM +/- LM + sobre crecimiento extremidad |
|
|
Síndrome Parkes Weber: CM + AVF + sobre crecimiento extremidad G |
|
|
Síndrome Servelle-Martorell: limb VM + sobre crecimiento óseo |
|
|
Síndrome Sturge-Weber: CM facial + leptomeningeo + anomalías oculares +/- hueso y/o sobre crecimiento de partes blandas G |
|
| CM de la extremidad+ hipertroia congenita non-progresiva de la extremidad | |
| Síndrome Maffucci: VM +/- hemangioma fusiforme+ encondroma | |
| Macrocefalia - CM (M-CM / MCAP) G | |
|
Microcefalia - CM (MICCAP) G |
|
|
Síndrome CLOVES: LM + VM + CM +/- AVM + sobre crecimiento lipomatosis G |
|
|
Síndrome Proteus: CM, VM y/o LM + sobre crecimiento asimétrico corporal G |
|
|
Sd Bannayan-Riley-Ruvalcaba: AVM + VM +macrocefalia, sobre crecimiento lipomatosis G |
|
| ANOMALÍAS VASCULARES PROVISIONALMENTE NO CLASIFICAS |
| Hemangioma Verrucoso |
| Angiokeratoma |
| Linfangioendoteliomatosis multifocal con trobocitopenia / angiomatosis cutaneovisceral con trombocitopenia (MLT/CAT) |
|
Linfangiomatosis Kaposiforme I (KLA ) |
| PTEN (tipo) hamartoma de partes blandas / 'angiomatosis' de partes blandas G |
Algunas lesiones pueden estare asociadas a trombocitopenia y/o coagulopatía de consumo. Ir a G para ver genética
| AVF | arteriovenous fistula |
| AVM | arteriovenous malformation |
| CAT | cutaneovisceral angiomatosis with thrombocytopenia |
| CAVM | capillary arteriovenous malformation |
| CCM | cerebral cavernous malformation |
| CLAVM | capillary lymphatic arteriovenous malformation |
| CLOVES | congenital lipomatous overgrowth, vascular malformations, epidermal nevi, skeletal/scoliosis and spinal abnormalities |
| CLM | capillary lymphatic malformation |
| CLVAVM |
capillary lymphatic venous arteriovenous malformation |
| CLVM | capillary lymphatic venous malformation |
| CM | capillary malformation |
| CM-AVM | capillary malformation-arteriovenous malformation |
| CMTC | cutis marmorata telangiectatica congenita |
| CNS | central nervous system |
| CVAVM | capillary venous arteriovenous malformation |
| CVM | capillary venous malformation |
| DIC | disseminated intravascular coagulopathy |
| GLA | generalized lymphatic anomaly |
| GSD | Gorham-Stout disease |
| GVM | glomuvenous malformation |
| HHT | hereditary hemorrhagic telangiectasia |
| HI | hemangioma of infancy / infantile hemangioma |
| IH | infantile hemangioma / hemangioma of infancy |
| INR | international normalized ratio |
| JPHT | juvenile polyposis hemorrhagic telangiectasia |
| KHE | kaposiform hemangioendothelioma |
| KLA | kaposiform lymphangiomatosis |
| KMP | Kasabach-Merritt phenomenon |
| LM | lymphatic malformation |
| LVM | lymphatic venous malformation |
| MCAP | megalencephaly-capillary malformation-polymicrogyria |
| M-CM | macrocephaly-capillary malformation |
| MICCAP | microcephaly-capillary malformation |
| MLT | Multifocal lymphangioendotheliomatosis with thrombocytopenia |
| NICH | non-involuting congenital hemangioma |
| PHACE | posterior fossa malformations, hemangioma, arterial anomalies, cardiovascular anomalies, eye anomalies |
| PILA |
papillary intralymphatic angioendothelioma |
| PICH | partially involuting congenital hemangioma |
| RICH | rapidly involuting congenital hemangioma |
| TA | tufted angioma |
| VM | venous malformation |
| VMCM | venous malformation cutaneo mucosal |
| MALFORMACIONES CAPILARES (CM) |
| CM Cutáneos y/o mucosos (mancha en vino de Oporto) GNAQ |
| CAM con hiperplasia de hueso y partes blandas |
| CM de CM-AVM RASA1 |
| Telangiectasia |
| Telangiectasia hemorràgica hereditaria (HHT) |
| HHT 1 ENG |
| HHT2 ACVRL1 |
| HHT3 |
| JPHT (telangiectasia juvenil poliposa hemorrágica ) SMAD4 |
| Otros |
| Telangiectasia congénita Cutis M armorata (CMTC) |
| Nevus simple / mancha Salmón |
| Otros |
| LYMPHATIC MALFORMATIONS (LM) |
| Linfedema Primario |
| Sindrome |
| Linfedema |
| Linfedema-Disitquiasis FOXC2 |
| Hipotricosis-linfedema-telangiectasia SOX18 |
| Linfedema Primario con mielodysplasia GATA2 |
| Anomalía linfática primaria generalizada ( Síndrome linfagiectasia-linfedema de Hennekam ) CCBE1 |
| Síndrome de microcefalia con o sin corioretinopatía , linfedema o retraso mental KIF11 |
| Linfedema - atresia coanal ( PTPN14) |
| MALFORMACIONES VENOSAS ( VM) |
| VM Común TIE2 somatic |
| VM cutáneo-mucosa Familiar (VMCM) TIE2 |
| VM Síndrome del nevo gomoso azul |
| Mlaformación Glomovenosa ( VM con células de glomus ) Glomulin |
| Malformación Cerebral cavernosa (CCM ) |
| CCM1 KRIT1 |
| CCM2 Malcavernin |
| CCM3 PDCD10 |
| MALFORMACIONES ARTERIOVENOSAS ( AVM) |
| Esporádico |
| En HHT |
| HHT1 ENG |
| HHT2 ACVRL1 |
| JPHT (telangiectasia juvenil poliposa hemorrágica ) SMAD4 |
| En CM-AVM RASA1 |
| FISTULAS ARTERIOVENOUS ( AVF) |
| Sporadic |
| In HHT |
| HHT1 ENG |
| HHT2 ACVRL1 |
| JPHT (juvenile polyposis hemorrhagic telangiectasia ) SMAD4 |
| In CM-AVM RASA1 |
| MALFORMACIONES VASCULARES ASOCIADAS A OTRAS ANOMALÍAS |
| Síndrome Klippel - Trenaunay |
| Síndrome Parkes Weber RASA1 |
| Síndrome Servelle-Martorell |
| Síndrome Sturge-Weber GNAQ |
| Limb CM + congenital non-progressive limb overgrowth |
| Síndrome Maffucci |
| Macrocefalia - CM ( M-CM or MCAP ) PIK3CA |
| Microcefalia - CM (MICCAP) STAMBP |
| Síndrome CLOVES PIK3CA |
| Síndrome Proteus AKT1 |
| Síndrome Bannayan-Riley-Ruvalcaba PTEN |
| ANOMALIAS VASCULARES PROVISIONALMENTE NO CLASIFICADAS) |
| Hemangioma Verrucoso |
| Linfangioendoteliomatosis Multifocal con trombocitopenia / angiomatosis cutáneovisceral con trombocitopenia ( MLT/CAT ) |
| Linfangiomatosis Kaposiforme I (KLA) |
| PTEN ( tipo ) hamartoma de partes blandas / "angiomatosis" de partes blandas ( PTEN ) |
Hemangioma Infantil
| PATRÓN |
|
- Focal - Multifocal - Segmentario - Indeterminado |
| TIPOS DIFERENTES |
|
- Superficial - Profundo - Mixto ( Superficial + profundo) - Reticular / abortivo / mínimo crecimiento - otros |
| ASOCIADO A OTRAS LESIONES | |
|
PHACE asociación / síndrome |
Malformaciones Fosa Posterior , Hemangioma, Anomalías Arteriales, Anomalías Cardiovasculares, Anomalías oculares, Fisura Esternal y/o rafe supraumbilical |
| LUMBAR ( SACRO, PELVIS ) asociación / síndrome | Hemangioma Corporal Inferior, Anomalías Urogenital, Ulceración , Mielopatía, Deformidades Ósea, Malformaciones Anorectales, Anomalías Arteriales y Anomalías Renales |
Anomalías vasculares posiblemente asociadas con nivel de plaquetas / alteraciones de la coagulación
| ANOMALÍAS | ALTERACIONES HEMATOLÓGICAS |
|
Angioma Encopetado Hemangioendotelioma Kaposiforme |
Trombocitopenia severa y sostenida con hipofibrinogenemia severa, coagulopatía de consumo y elevación del dímero D ( fenómeno de Kasabach-Merritt) |
| Hemangioma Congenital Rápidamente involutivo | Trombocitopenia transitoria leve-moderada, +/- coagulopatía de consumo y elevado dímero D |
| Coagulopatía intravascular crónica localizada con elevación del dímero D, +/- hipofibrinogenemia, y +/- trombocitopenia moderada. ( Puede progresar a la CID después de un traumatismo o una operación ) | |
| Malformaciones Linfáticas | Coagulopatía intravascular crónica localizada con elevación del dímero D y +/- trombocitopenia leve a moderada ( Considerar Linfangiomatosis kaposiforme ) ( Puede progresar a CID después de una traumatismo o una operación ) |
| Lifangioendoteliomatosis | Sustained, fluctuating, moderate to profound thrombocytopenia with gastrointestinal tract |
|
Multifocal con trombocitopenia Angiomatosis Cutáneovisceral con trombocitopenia |
bleeding or pulmonary hemorrhage |
| Linfangiomatosis Kaposiforme |
Trombocitopenia de moderada a severa sostenida, fluctuante , con sangrado del tracto gastrointestinal o hemorragia pulmonar. Trombocitopenia leve a moderada, +/- hipofibrinogenemia , elevación dímero D |